2 快速入门
本章将介绍DS-PAW的各种功能的基本使用,具体包括: 结构弛豫计算 、 自洽计算 、 能带(投影能带)计算 、 态密度(投影态密度)计算 、 势函数计算 、 电子局域密度计算 、 部分电荷密度计算 、 杂化泛函计算 、 范德瓦尔斯修正计算 、 偶极修正计算 、 DFT+U计算 、 背景电荷计算 、 光学性质计算 、 频率计算 、 弹性常数计算 、 过渡态计算 、 声子谱计算 、 自旋轨道耦合计算 、 分子动力学模拟 、 外加电场计算 、 铁电计算 、 bader电荷计算 、 能带反折叠计算 、 介电常数计算 、 压电张量计算 、 固定基矢弛豫计算 、 声子热力学性质计算 、 solid state neb计算 、 溶剂化能计算 、 固定电势计算 、 wannier插值能带计算 ;DS-PAW软件的参数大致可分为以下几类:与物理结构相关的参数、与计算性质相关的参数、与计算精度相关的参数、与收敛相关的参数,基本参数多有默认值。 本章筛选部分参数进行介绍,参数列表及详情另见 参数说明 部分 。
2.1 relax结构弛豫计算
在密度泛函理论(DFT)中结构弛豫指的改变初始的结构的晶胞及原子位置从而优化得到总能量的局域最小值。 通过结构弛豫计算,可以减少在每个原子上的受力,从而得到较为稳定的结构(一定程度上,可以通过计算声子谱或者频率来验证结构的稳定性)。 使用建模软件搭建的结构一般原子受力会比较大,且即便是其他DFT软件优化过的结构,在另外一个DFT计算软件中原子受力也不一定是最小的,因此在计算某个结构的具体性质之前需进行结构弛豫计算。
\(Si\) 原子结构弛豫输入文件
输入文件包含参数文件 relax.in 和结构文件 structure.as , relax.in 如下:
1# task type
2task = relax
3#system related
4sys.structure = structure.as
5sys.symmetry = true
6sys.functional = PBE
7sys.spin = none
8#scf related
9cal.methods = 2
10cal.smearing = 1
11cal.ksamping = G
12cal.kpoints = [10, 10, 10]
13cal.cutoffFactor = 1.5
14
15#relax related
16relax.max = 60
17relax.freedom = atom
18relax.convergenceType = force
19relax.convergence = 0.05
20relax.methods = CG
21
22io.wave = false
23io.charge = false
relax.in 文件大致可以分为4部分参数:
第一部分指定计算类型,由task参数控制:
task:设置计算类型,本次计算为relax即结构弛豫;
第二部分指定系统相关参数,系统相关参数以sys.开头,一般是一些与体系的结构、泛函、磁性、对称性相关的参数:
sys.structure:指定体系的结构文件,DS-PAW支持 .as 和 .h5 的结构文件格式(早期json文件可支持但不建议用户使用,后续DS-PAW发布版本将会完全摒弃json格式的输出), .as 文件可使用Device Studio软件直接生成,也可手动构造;sys.symmetry:设置DS-PAW计算时是否需要使用对称性;sys.functional:设置泛函,目前程序支持 LDA 、 PBE 及多种修正泛函;sys.spin:设置体系的磁性,由于 Si 没有磁性,因此将sys.spin设置为 none ;
第三部分指定计算相关的参数,这类参数以cal.开头:
cal.methods: 设置自洽电子步优化方法,2表示使用 Residual minimization 方法;cal.smearing:设置每个波函数的部分占有数方法,1表示使用 Gaussian smearing 方法;cal.ksamping:自动生成布里渊区 k点 网格方法,G表示使用 Gamma centered 方法;cal.kpoints: 设置布里渊区 k点 网格取样大小,一般K点的大小需要根据体系晶格的大小和体系的周期性来设置;
第四部分指定结构弛豫相关参数,例如结构弛豫的方法、结构弛豫的类型、结构弛豫的精度等相关参数,结构弛豫指的是优化原子位置得到总能量的局域最小值的结构,一般也称之为离子步优化;
relax.max:设置结构驰豫时,最大的离子步步数;relax.freedom:设置结构驰豫的自由度,atom表示只弛豫原子位置;另有可选值volume表示只弛豫晶格体积;all表示弛豫弛豫原子位置、晶胞形状和体积;relax.convergenceType: 设置结构弛豫收敛的判据类型,force表示以原子受力作为判据,另有可选值energy;relax.convergence:设置结构驰豫时,原子受力的收敛精度大小;relax.methods:设置结构驰豫的方法,CG表示共轭梯度法;
structure.as 文件参考如下:
1Total number of atoms
22
3Lattice
40.00 2.75 2.75
52.75 0.00 2.75
62.75 2.75 0.00
7Direct
8Si -0.115000000 -0.125000000 -0.125000000
9Si 0.125000000 0.125000000 0.125000000
structure.as 文件结构固定,必须严格对照每一行写入相应信息:
第一行为固定提示行
第二行为总的原子个数
第三行为固定提示行
第四至六行为晶胞信息
第七行为原子坐标的形式,可选值为 Direct 和 Cartesian (全拼且首字母必须为大写)
第八行开始写入原子坐标信息,每一行的开头必须标注坐标所描述的原子的名称
为展示结构弛豫前后结构的变化,此例手动将第一个Si原子在x方向的坐标从 -0.125 改为 -0.115 。
备注
如果想要固定原子,则在第7行加入Fix_x Fix_y Fix_z标签,然后在每个原子对应的位置加入F或T,F表示不固定,T表示固定。
1Direct Fix_x Fix_y Fix_z
2Si -0.115000000 -0.125000000 -0.125000000 F F F
3Si 0.125000000 0.125000000 0.125000000 T T T
run程序运行
准备好输入文件之后,将 relax.in 和 structure.as 文件上传到安装了DS-PAW的环境上,这里将以linux环境为例子进行介绍;
在无图形界面的linux环境下运行软件的方式与在windows的环境下运行程序会有很大的区别,在linux下需要通过命令行的方式来运行程序;
一般情况下需要先加载一下环境变量,通常会将需要用的环境变量写入文本或者 ~/.bashrc ,通过 source 的命令来加载环境;
环境加载完成之后,运行 DS-PAW relax.in ,此时使用单机版的DS-PAW进行计算;如需并行计算则需运行 DS-PAW -mpi mpirun -mpiargs “-n 2” relax.in ,-mpi 指定mpirun的名称,
-mpiargs 指定mpirun后面的参数。命令介绍见第一节 软件简介 部分。
使用排队系统(例如PBS、slurm等)提交任务需先配置相应的 .pbs 或 .slurm 脚本,使用 qsub DS-PAW.pbs 或 sbatch DS-PAW.slurm 提交任务即可。
analysis计算结果分析
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 relax.h5 、 latestStructure.as 等输出文件:
DS-PAW.log :DS-PAW计算之后得到的日志文件;
relax.h5 :弛豫计算对应的h5输出文件,结构解析见 输出文件格式说明 部分,DS-PAW可读取该h5文件进行续算;
latestStructure.as :弛豫终点的as结构文件,可直接查看数据;
将 latestStructure.as 拖入Device Studio查看结构如下所示:
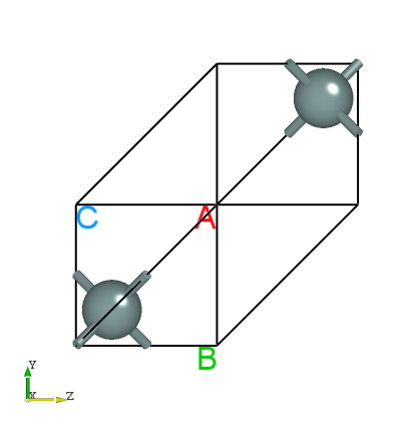
查看 latestStructure.as 文件,可得弛豫结束后的晶胞信息如下:
1Total number of atoms
22
3Lattice
40.0000000000000000 2.7500000000000000 2.7500000000000000
52.7500000000000000 0.0000000000000000 2.7500000000000000
62.7500000000000000 2.7500000000000000 0.0000000000000000
7Direct
8Si 0.8801735223171917 0.8748246492235915 0.8748246492235915
9Si 0.1298264776828063 0.1251753507764085 0.1251753507764085
本次结构弛豫计算进行了3个离子步的计算,弛豫结束的构型中,手动移动的第一个Si原子在x方向上的坐标在弛豫计算之后完成校正。
备注
单机版DS-PAW运行命令为软件名+输入文件名,如果你的输入文件名为abc.in那么只要执行DS-PAW abc.in即可。
该弛豫计算的收敛判据选取为原子受力,若以能量作为收敛判据,可以设置relax.covergenceType = energy。
2.2 scf自洽计算
自洽计算能得到特定晶体的电荷密度和波函数文件,电荷密度文件用于后续计算该体系的能带、态密度等电子结构性质。 特别需要注意是:自洽与能带、态密度等电子结构性质计算是有先后顺序的,必须先进行自洽计算得到电荷密度才能进一步计算能带、态密度等电子结构性质。
\(Si\) 原子自洽计算之准备输入文件
输入文件包含参数文件 scf.in 和结构文件 structure.as , scf.in 如下:
1# task type
2task = scf
3#system related
4sys.structure = structure.as
5sys.symmetry = true
6sys.functional = PBE
7sys.spin = none
8#scf related
9cal.methods = 2
10cal.smearing = 1
11cal.ksamping = G
12cal.kpoints = [10, 10, 10]
13cal.cutoffFactor = 1.5
14#outputs
15io.charge = true
16io.wave = true
scf.in 输入自洽相关参数介绍:
task:设置计算类型,此次计算为 scf 自洽计算;cal.cutoffFactor:设置cal.cutoff的系数,计算采用的截断能大小等于cal.cutoff*cal.cutoffFactor;io.charge:控制电荷密度文件输出的开关;io.wave:控制波函数文件输出的开关;
structure.as 文件参考如下:
1Total number of atoms
22
3Lattice
40.00 2.75 2.75
52.75 0.00 2.75
62.75 2.75 0.00
7Direct
8Si -0.125000000 -0.125000000 -0.125000000
9Si 0.125000000 0.125000000 0.125000000
常规自洽计算会选取结构弛豫得到的稳态构型作为结构输入;
备注
在结构弛豫和自洽计算中可保存elf、potential的数据,只需要将io.elf和io.potential设置为true即可;
计算时如需给体系添加背景电荷,可直接设置sys.electron参数,该参数指定价电子的总数。
run程序运行
准备好输入文件 scf.in 和 structure.as 后,将文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW scf.in 。
analysis计算结果分析
根据上述的输入文件,计算完成之后可得到 DS-PAW.log 、 scf.h5 、 rho.bin 、 wave.bin 、 rho.h5 等输出文件。
DS-PAW.log :DS-PAW计算之后得到的日志文件,记录自洽计算中能量迭代等主要信息;
scf.h5 :自洽计算对应的h5输出文件,结构解析见 输出文件格式说明 部分;
rho.bin :电荷密度的二进制文件,用于后续的后处理计算;
rho.h5 :电荷密度的 h5 格式文件,可简单转换成VESTA能读取的格式(见 辅助工具使用教程 ),从而将电荷密度信息可视化;
wave.bin :波函数的二进制文件,用于后续计算;
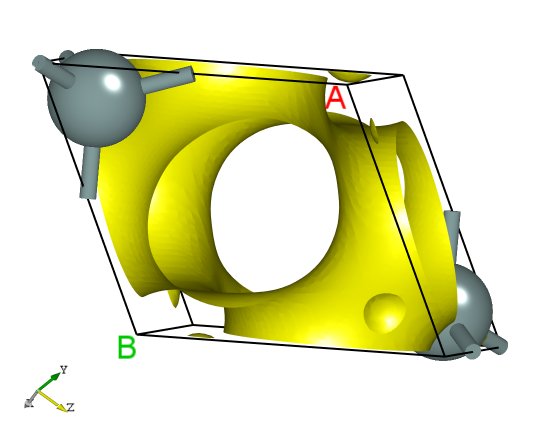
可通过python脚本将 rho.h5 文件转成 VESTA 软件支持的格式,具体操作见 辅助工具使用教程 部分。处理可得一维、二维、三维电荷密度图,其中三维图效果应如下所示:
2.3 band能带计算
普通能带计算有两种方式可完成,task=band 的两步法及 task=scf 的一步法。此节以Si体系为例,介绍两种方法下对应的参数设置。
\(Si\) 体系能带计算输入文件
task = band 两步算
输入文件包含参数文件 scf.in 和 band.in ,结构文件 structure.as , scf.in 设置与上节自洽计算一致,band.in 参数如下:
1# task type
2task = band
3#system related
4sys.structure = structure.as
5sys.symmetry = true
6sys.functional = PBE
7sys.spin = none
8
9cal.iniCharge = ./rho.bin
10cal.methods = 2
11cal.smearing = 1
12cal.cutoffFactor = 1.5
13cal.totalBands = 12
14
15#band related
16band.kpointsLabel= [G,X,W,K,G,L]
17band.kpointsCoord= [0, 0, 0, 0.5, 0, 0.5, 0.5, 0.25, 0.75, 0.375, 0.375, 0.75, 0, 0, 0, 0.5, 0.5, 0.5]
18band.kpointsNumber= [30, 30, 30, 30, 30]
band.in 输入参数介绍:
在能带计算中可以尽量保留 sys. 和 cal. 的参数到 band.in 中,之后设置能带计算特有的参数即可:
task:设置计算类型,本次计算为band能带计算;cal.iniCharge:设置电荷密度文件的路径,支持绝对路径及相对路径,这里./表示当前路径下的 rho.bin 文件;
能带计算中新增了一部分能带相关的参数,这些参数只在能带计算中起作用:
band.kpointsLabel:设置能带计算时高对称点标签,一个band.kpointsCoord对应一个band.kpointsLabel;band.kpointsCoord:设置能带计算时高对称点的分数坐标,每三个数为一组;band.kpointsNumber:设置每相邻两个高对称点间的k点个数,有两种写法:当设置参数为 band.kpointsNumber= [30, 30, 30, 30, 30] 时,所有高对称点之间撒点数均为30;
当设置参数为 band.kpointsNumber= [30] 时,高对称点G与X之间撒点数为30,以此求得撒点密度;对高对称点X与W、W与K、K与G、G与L之间进行等密度撒点,实际撒点数可从DS-PAW.log的参数打印部分获取;
band.EfShift:决定是否读取rho.bin中的EFermi作为band计算输出中的EFermi。默认为true,表示从rho.bin读取EFermi。
structure.as 文件同自洽计算。(见2.2节)
备注
两步算时,scf.in 和 band.in 中参数 cal.cutoffFactor及cal.cutoff 必须保持一致,否则会出现格点数据不匹配的问题。
cal.iniCharge指定自洽计算生成的电荷密度文件 rho.bin 所在路径。
task = scf 一步算
输入文件包含参数文件 scf.in ,结构文件 structure.as , scf.in 参数如下:
1# task type
2task = scf
3#system related
4sys.structure = structure.as
5sys.symmetry = true
6sys.functional = PBE
7sys.spin = none
8#scf related
9cal.methods = 2
10cal.totalBands = 12
11cal.smearing = 1
12cal.ksamping = G
13cal.kpoints = [10, 10, 10]
14cal.cutoffFactor = 1.5
15#outputs
16io.charge = true
17io.wave = true
18#band related
19io.band=true
20band.kpointsLabel= [G,X,W,K,G,L]
21band.kpointsCoord= [0, 0, 0, 0.5, 0, 0.5, 0.5, 0.25, 0.75, 0.375, 0.375, 0.75, 0, 0, 0, 0.5, 0.5, 0.5]
22band.kpointsNumber= [30, 30, 30, 30, 30]
备注
能带一步算对应结果文件为 scf.h5 ,此时能带数据存储在 scf.h5 文件中,可直接调用 辅助工具使用教程 的 bandplot.py 脚本处理 scf.h5 文件。
io.band=true 只在 task=scf 时生效。
当io.band生效时,不再需要设置cal.iniCharge = ./rho.bin,对K空间高对称点的求解将在scf计算时同步完成。
scf.in文件中需给出两类k点,cal.kpoints给出自洽计算的k点,band.kpoints相关参数给出能带计算的k点,两部分k点缺一不可。
run程序运行
以两步算为例,将参数控制文件 scf.in 、 band.in 和结构文件 structure.as 上传到服务器,按照结构弛豫中介绍的方法依次执行 DS-PAW scf.in 、 DS-PAW band.in 。
analysis计算结果分析
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 scf.h5 、 band.h5 等输出文件。
DS-PAW.log :DS-PAW能带计算之后得到的日志文件,可直接读取带隙、VBM、CBM等重要信息;
band.h5 :能带计算对应的 h5 输出文件;保存能量本征值等重要数据,具体的数据结构详见 输出文件格式说明 部分;
可使用 python 对 band.h5 进行数据处理,具体操作见 辅助工具使用教程 部分。处理得到的能带图效果应如下所示:

备注
能带计算一步法和两步法得到的能带图效果一致。
2.4 pband投影能带计算
投影能带是指在能带计算过程中将每条能带每个K点上的能量展开为每个原子及其轨道的贡献。
\(Si\) 投影能带计算输入文件
投影能带的输入文件包含参数文件 pw_band.in 结构文件 structure.as 和自洽计算得到的二进制电荷密度文件 rho.bin , pw_band.in 如下:
1# task type
2task = band
3#system related
4sys.structure = structure.as
5sys.symmetry = true
6sys.functional = PBE
7sys.spin = none
8
9cal.iniCharge = ./rho.bin
10cal.methods = 2
11cal.smearing = 1
12cal.cutoffFactor = 1.5
13cal.totalBands = 12
14
15#band related
16band.kpointsLabel= [G,X,W,K,G,L]
17band.kpointsCoord= [0, 0, 0, 0.5, 0, 0.5, 0.5, 0.25, 0.75, 0.375, 0.375, 0.75, 0, 0, 0, 0.5, 0.5, 0.5]
18band.kpointsNumber= [30, 30, 30, 30, 30]
19band.project=true
pw_band.in 输入参数介绍:
投影能带计算与普通能带的区别在于在计算参数中设置了 band.project 参数:
band.project :控制能带计算中投影计算的开关;
run程序运行
准备好输入文件 pw_band.in 和 structure.as 以及 rho.bin 之后,将文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW pw_band.in 。
analysis计算结果分析
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 band.h5 等输出文件。
DS-PAW.log :DS-PAW能带计算之后得到的日志文件;
band.h5 :能带计算对应的 h5 输出文件,此时投影能带的数据也将会被保存在 band.h5 中,具体的数据结构详见 输出文件格式说明 部分;
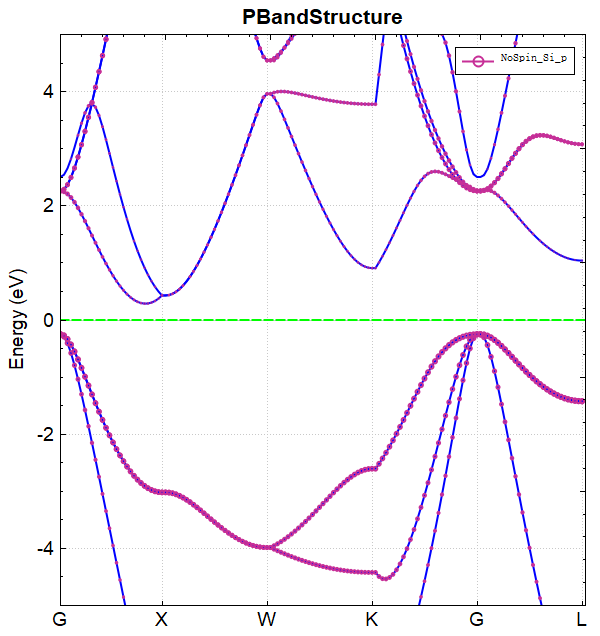
可使用 python 对 band.h5 进行数据处理,具体操作见 辅助工具使用教程 部分。处理得到的能带图效果应如下所示:
2.5 dos态密度计算
态密度计算有两种方式可完成,task=dos 的两步法及 task=scf 的一步法。此节以Si体系为例,介绍两种方法下对应的参数设置。
\(Si\) 体系态密度计算输入文件
task = dos 两步算
输入文件包含参数文件 scf.in 、 dos.in 及结构文件 structure.as , scf.in 设置与自洽计算一致,dos.in 参数如下:
1# task type
2task = dos
3#system related
4sys.structure = structure.as
5sys.symmetry = true
6sys.functional = PBE
7sys.spin = none
8
9cal.iniCharge = ./rho.bin
10cal.methods = 2
11cal.smearing = 4
12cal.ksamping = G
13cal.kpoints = [20, 20, 20]
14cal.cutoffFactor = 1.5
15
16#dos related
17dos.range=[-10, 10]
18dos.resolution=0.05
dos.in 输入参数介绍:
在态密度计算中可以尽量保留 sys. 和 cal. 的参数到 dos.in 中,之后设置态密度计算特有的参数即可:
task:设置计算类型,本次计算为dos态密度计算;cal.iniCharge:设置电荷密度的读取路径,支持绝对路径及相对路径,这里./表示当前路径下的 rho.bin 文件;cal.kpoints:设置k点网格密度,态密度计算时k点建议增大到自洽计算的两倍左右;
态密度计算中新增了一部分态密度相关的参数,这些参数只在态密度计算中起作用:
dos.range:设置态密度计算时能量的区间;dos.resolution:设置态密度计算能量间隔精度,态密度计算的点数即为 dos.range 的差值与 dos.resolution 的比值+1;
structure.as 文件同自洽计算。(见2.2节)
备注
两步算时,scf.in 和 dos.in 中参数 cal.cutoffFactor及cal.cutoff 必须保持一致,否则会出现格点数据不匹配的问题。
task = scf 一步算
输入文件包含参数文件 scf.in ,结构文件 structure.as , scf.in 参数如下:
1# task type
2task = scf
3#system related
4sys.structure = structure.as
5sys.symmetry = true
6sys.functional = PBE
7sys.spin = none
8#scf related
9cal.methods = 2
10cal.smearing = 4
11cal.ksamping = G
12cal.kpoints = [10, 10, 10]
13cal.cutoffFactor = 1.5
14#outputs
15io.charge = true
16io.wave = true
17#dos related
18io.dos=true
19dos.range=[-10, 10]
20dos.resolution=0.05
备注
态密度一步算对应结果文件为 scf.h5 ,此时态密度数据存储在 scf.h5 文件中,可直接调用 辅助工具使用教程 的 dosplot.py 脚本处理 scf.h5 文件。
io.dos=true 只在 task=scf 时生效。
当io.dos生效时,不再需要设置cal.iniCharge = ./rho.bin,此时通过自洽计算获得DOS。
run程序运行
以两步算为例,将参数控制文件 scf.in 、 dos.in 和结构文件 structure.as 上传到服务器,按照结构弛豫中介绍的方法依次执行 DS-PAW scf.in 、 DS-PAW dos.in 。
analysis计算结果分析
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、scf.h5 、 dos.h5 等输出文件。
DS-PAW.log :DS-PAW态密度计算之后得到的日志文件;
dos.h5 :态密度计算对应的h5文件,具体结构见 输出文件格式说明 部分 ;
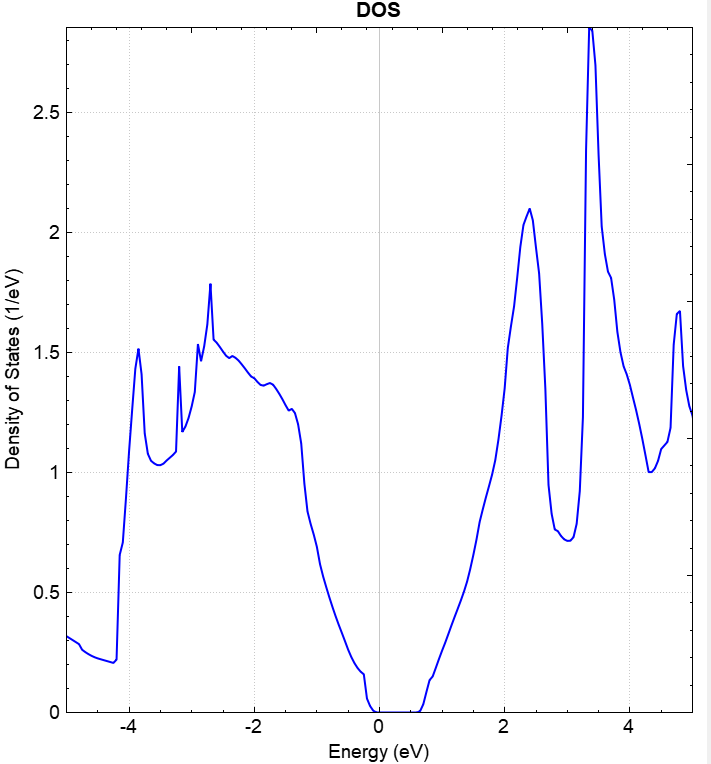
可使用 python 对 dos.h5 进行数据处理,具体操作见 辅助工具使用教程 部分。处理得到的态密度图效果应如下所示:
2.6 pdos投影态密度计算
投影态密度的计算是指在态密度计算过程中将态每个能量下的态密度展开为每个原子及其轨道的态密度贡献。
\(Si\) 投影态密度计算输入文件
投影态密度的输入文件包含参数文件 pdos.in 结构文件 structure.as 和自洽计算得到的电荷密度文件 rho.bin , pdos.in 如下:
1# task type
2task = dos
3#system related
4sys.structure = structure.as
5sys.symmetry = true
6sys.functional = PBE
7sys.spin = none
8
9cal.iniCharge = ./rho.bin
10cal.methods = 2
11cal.smearing = 4
12cal.ksamping = G
13cal.kpoints = [20, 20, 20]
14cal.cutoffFactor = 1.5
15
16#dos related
17dos.range=[-10, 10]
18dos.resolution=0.05
19dos.project = true
pdos.in 输入参数介绍:
投影态密度与普通态密度的区别在于在计算参数中设置了 dos.project 参数:
dos.project:控制态密度计算中投影计算的开关;
run程序运行
准备好输入文件 pdos.in 和 structure.as 以及 rho.bin 之后,将文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW pdos.in 。
analysis计算结果分析
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 dos.h5 等输出文件。
DS-PAW.log :DS-PAW态密度计算之后得到的日志文件;
dos.h5 :态密度计算对应的 h5 输出文件;投影态密度的数据保存在 dos.h5 文件中,具体的数据结构详见 输出文件格式说明 部分;
可使用 python 对 dos.h5 进行数据处理,具体操作见 辅助工具使用教程 部分。处理得到的投影态密度图效果应如下所示:
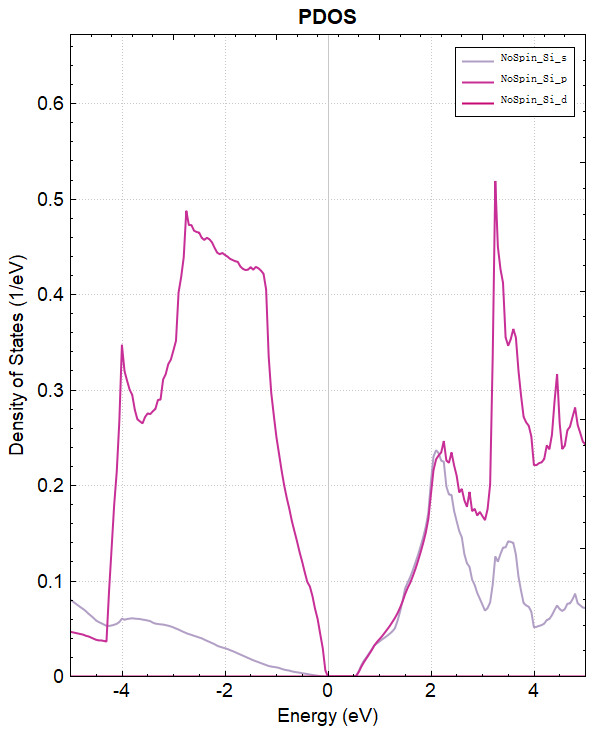
2.7 potential势函数计算
势函数计算有两种方式可完成,task=potential 的两步法及 task=scf 的一步法。此节以Si体系为例,介绍两种方法下对应的参数设置。
\(Si\) 势函数计算输入文件
task = potential 两步算
输入文件包含参数文件 scf.in 、 potential.in 和结构文件 structure.as , scf.in 设置与自洽计算一致, potential.in 设置如下:
1# task type
2task = potential
3#system related
4sys.structure = structure.as
5sys.symmetry = true
6sys.functional = PBE
7sys.spin = none
8
9cal.iniCharge = ./rho.bin
10cal.methods = 2
11cal.smearing = 1
12cal.ksamping = G
13cal.kpoints = [10, 10, 10]
14cal.cutoffFactor = 1.5
15
16#potential related
17potential.type=all
potential.in 输入参数介绍:
在势函数计算中可以尽量保留 sys. 和 cal. 的参数到 potential.in 中,之后设置势函数计算特有的参数即可:
task:设置计算类型,本次计算为potential势函数计算;cal.iniCharge:设置读取电荷密度文件的路径,支持绝对路径及相对路径,这里./表示当前路径下的 rho.bin 文件;
势函数计算中新增参数:
potential.type:控制势函数保存的类型,当选择 all 的时候,势函数计算完成之后会同时保存静电势(离子势和hartree势之和)及局域势(静电势和交换关联势之和);
structure.as 文件同自洽计算。(见2.2节)
备注
两步算时,scf.in 和 potential.in 中参数 cal.cutoffFactor及cal.cutoff 必须保持一致,否则会出现格点数据不匹配的问题。
如果所计算的体系需要考虑偶极修正,此时用户需要在自洽和势函数计算文件中都加入corr.dipol = true以及corr.dipolDirection参数,corr.dipol = true表示打开偶极修正的开关,corr.dipolDirection表示设置偶极修正的方向a、b、c分别表示沿着晶格矢量的a、b、c方向。
偶极修正的具体案例见应用案例:Au-Al体系功函数计算案例。
task = scf 一步算
输入文件包含参数文件 scf.in ,结构文件 structure.as , scf.in 参数如下:
1# task type
2task = scf
3#system related
4sys.structure = structure.as
5sys.symmetry = true
6sys.functional = PBE
7sys.spin = none
8#scf related
9cal.methods = 2
10cal.smearing = 1
11cal.ksamping = G
12cal.kpoints = [10, 10, 10]
13cal.cutoffFactor = 1.5
14#outputs
15io.charge = true
16io.wave = true
17#potential related
18io.potential = true
备注
势函数一步算对应结果文件为 scf.h5 ,此时势函数数据存储在 scf.h5 文件中,可直接调用 辅助工具使用教程 的势函数处理脚本分析 scf.h5 文件。
io.potential=true 只在 task=scf 时生效。
run程序运行
以两步算为例,将参数控制文件 scf.in 、 potential.in 和结构文件 structure.as 上传到服务器,按照结构弛豫中介绍的方法依次执行 DS-PAW scf.in 、 DS-PAW potential.in 。
analysis计算结果分析
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 scf.h5 、 potential.h5 等输出文件。
DS-PAW.log :DS-PAW势函数计算之后得到的日志文件;
potential.h5 :势函数计算对应的 h5 输出文件,具体结构见 输出文件格式说明 部分;
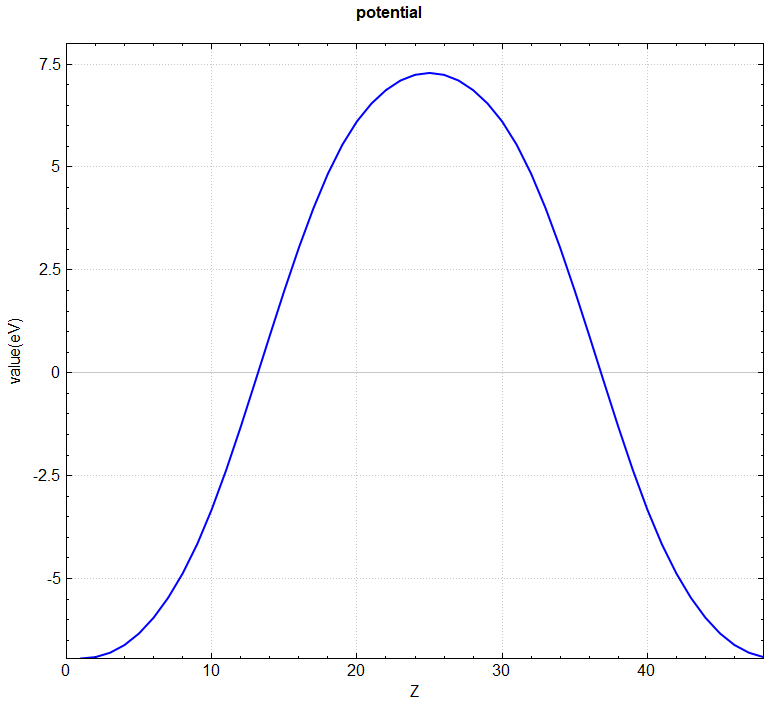
可使用 python 脚本将 potential.h5 格式的转化成 VESTA 软件支持的格式,也可直接使用脚本对三维势函数进行面内平均,具体操作见 辅助工具使用教程 部分。处理得到的真空方向势函数曲线如下所示:
2.8 elf电子局域密度计算
电子局域密度计算有两种方式可完成,task=elf 的两步法及 task=scf 的一步法。此节以Si体系为例,介绍两种方法下对应的参数设置。
\(Si\) 电子局域密度计算输入文件
task = elf 两步算
输入文件包含参数文件 scf.in 和 ELF.in ,结构文件 structure.as , scf.in 设置与自洽计算一致, ELF.in 设置如下:
1# task type
2task = elf
3#system related
4sys.structure = structure.as
5sys.symmetry = true
6sys.functional = PBE
7sys.spin = none
8
9cal.iniCharge = ./rho.bin
10cal.methods = 2
11
12cal.smearing = 1
13cal.ksamping = G
14cal.kpoints = [10, 10, 10]
15cal.cutoffFactor = 1.5
ELF.in 输入参数介绍:
在ELF计算中可以尽量保留 sys. 和 cal. 的参数到 ELF.in 中:
task:设置计算类型,本次计算为ELF计算;cal.iniCharge:设置电荷密度文件的读取路径,支持绝对路径及相对路径,这里./表示当前路径下的 rho.bin 文件;
structure.as 文件同(见2.2节)自洽计算的。
备注
两步算时,scf.in 和 ELF.in 中参数 cal.cutoffFactor及cal.cutoff 必须保持一致,否则会出现格点数据不匹配的问题。
ELF计算不支持non-collinear计算。
task = scf 一步算
输入文件包含参数文件 scf.in ,结构文件 structure.as , scf.in 参数如下:
1# task type
2task = scf
3#system related
4sys.structure = structure.as
5sys.symmetry = true
6sys.functional = PBE
7sys.spin = none
8#scf related
9cal.methods = 2
10cal.smearing = 1
11cal.ksamping = G
12cal.kpoints = [10, 10, 10]
13cal.cutoffFactor = 1.5
14#outputs
15io.charge = true
16io.wave = true
17#elf related
18io.elf = true
备注
电子局域密度一步算对应结果文件为 scf.h5 ,此时电子局域密度数据存储在 scf.h5 文件中,可直接调用 辅助工具使用教程 的电子局域密度处理脚本分析 scf.h5 文件。
io.elf=true 只在 task=scf 时生效。
ELF计算不支持non-collinear计算。
run程序运行
以两步算为例,将参数控制文件 scf.in 、 ELF.in 和结构文件 structure.as 上传到服务器,按照结构弛豫中介绍的方法依次执行 DS-PAW scf.in 、 DS-PAW ELF.in 。
analysis计算结果分析
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 scf.h5 、 elf.h5 等输出文件。
DS-PAW.log :DS-PAW局域密度计算之后得到的日志文件;
elf.h5 :ELF计算对应的 h5 输出文件, 具体结构见 输出文件格式说明 部分;
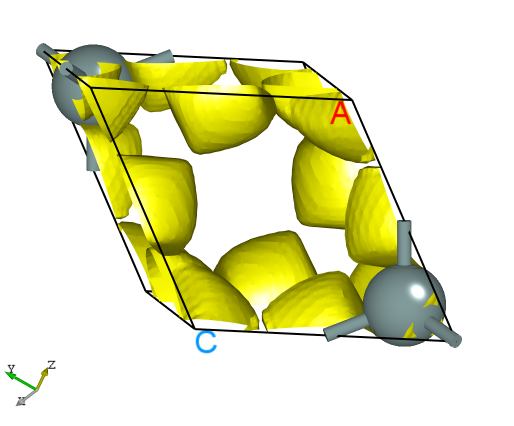
可使用 python 脚本将 elf.h5 格式的转化成 VESTA 软件支持的格式,具体操作见 辅助工具使用教程 部分。处理得到的三维电子局域密度图效果应如下所示:
2.9 pcharge部分电荷密度计算
本节将以石墨烯为例分析指定k点下特定能带的电荷密度,自洽完成之后准备部分电荷密度的计算,并对部分电荷密度作图进行分析。
graphene石墨烯部分电荷密度计算输入文件
输入文件包含参数文件 pcharge.in 和结构文件 structure.as ,自洽计算得到的电荷密度文件 rho.bin 和波函数文件 wave.bin , pcharge.in 如下:
1# task type
2task = pcharge
3#system related
4sys.structure = structure.as
5sys.symmetry = true
6sys.functional = PBE
7sys.spin = none
8
9cal.methods = 2
10cal.smearing = 1
11cal.ksamping = G
12cal.kpoints = [9, 9, 1]
13cal.cutoffFactor = 1.5
14cal.iniCharge = ./rho.bin
15cal.iniWave = ./wave.bin
16
17#pcharge related
18pcharge.bandIndex = [4,5]
19pcharge.kpointsIndex = [12]
20pcharge.sumK= false
pcharge.in 输入参数介绍:
在部分电荷密度计算中可以尽量保留sys.和cal.的参数到 pcharge.in 中,之后设置部分电荷密度计算特有的参数即可:
task: 设置计算类型,本次计算为部分电荷密度计算;cal.iniCharge: 设置电荷密度文件的读取路径,支持绝对路径及相对路径,这里./表示当前路径下的 rho.bin 文件;cal.iniWave: 设置波函数文件的读取路径,支持绝对路径及相对路径,这里./表示当前路径下的 wave.bin 文件;pcharge.bandIndex: 设置需进行电荷密度分析的能带序号,这里[4,5]表示分析能带4和能带5的电荷密度;pcharge.kpointsIndex: 设置进行电荷密度分析的K点序号,这里[12]表示分析两条能带的电荷密度时k点序号都为12;pcharge.sumK: 控制是否将所分析的所有K点能带数据相加。这里false表示不相加;
structure.as 文件参考如下:
1Total number of atoms
22
3Lattice
42.46120000 0.00000000 0.00000000
5-1.23060000 2.13146172 0.00000000
60.00000000 0.00000000 6.70900000
7Cartesian
8C 0.61530000 0.35524362 3.35450000
9C 0.61530000 1.77621810 3.35450000
备注
部分电荷密度分两步完成,第二步必须读取自洽计算的电荷密度文件rho.bin及波函数文件wave.bin。
run程序运行
准备好输入文件 pcharge.in 、 structure.as 以及自洽计算得到的 rho.bin 、 wave.bin 文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW pcharge.in 。
analysis计算结果分析
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、pcharge.h5 等输出文件。
DS-PAW.log :DS-PAW部分电荷密度计算之后得到的日志文件;
pcharge.h5 :部分电荷密度计算完成之后的h5数据文件,此时两条能带的电荷密度数据被保存在 pcharge.h5 中,具体的数据结构详见 输出文件格式说明 部分;
可使用 python 对 pcharge.h5 进行数据处理,具体操作见 辅助工具使用教程 部分。处理k点序号为 12 时能带 4 的电荷密度图效果应如下所示:

2.10 hse杂化泛函计算
本节将以Si体系为例,介绍DS-PAW程序通过自洽中直接计算能带的方法计算杂化泛函能带,观察使用杂化泛函计算后能带带隙的变化。
\(Si\) 杂化泛函计算输入文件
输入文件包含参数文件 ioband.in 和结构文件 structure.as , ioband.in 如下:
1# task type
2task = scf
3#system related
4sys.structure = structure.as
5sys.symmetry = true
6sys.spin = none
7#scf related
8cal.methods = 1
9cal.totalBands = 12
10cal.smearing = 1
11cal.ksamping = G
12cal.kpoints = [5, 5, 5]
13cal.cutoffFactor = 1.5
14#band related
15io.band = true
16band.kpointsCoord=[0.62500000,0.25000000,0.62500000,0.50000000,0.00000000,0.50000000,0.00000000,0.00000000,0.00000000,0.50000000,0.00000000,0.50000000,0.50000000,0.25000000,0.75000000,0.37500000,0.37500000,0.75000000,0.00000000,0.00000000,0.00000000]
17band.kpointsLabel = [U,X,G,X,W,K,G]
18band.kpointsNumber = [20,20,20,20,20,20]
19band.project = false
20#HSE related
21sys.hybrid=true
22sys.hybridType=HSE06
23#outputs
24io.charge = true
25io.wave = true
ioband.in 输入参数介绍:
在杂化泛函计算中可以尽量保留sys.和cal.的参数到 ioband.in 中,之后设置杂化泛函计算特有的参数即可:
sys.hybrid: 控制杂化泛函计算的开关,true表示引入杂化泛函计算;sys.hybridType: 设置杂化泛函的类型,此例为HSE06;
structure.as 文件同自洽计算。(见2.2节)
备注
不同于普通计算使用 sys.functional 设置泛函类型,杂化泛函计算通过参数 sys.hybridType 来控制杂化泛函种类。
杂化泛函计算只支持 task=scf/relax 的计算,因此杂化泛函能带计算只能通过一步算完成。
杂化泛函计算建议使用 damped MD/conjugated gradient 方法进行电子自洽计算,对应参数设置为 cal.methods = 4/5
杂化泛函计算也可使用 block Davidson 方法进行电子自洽计算,即此例所设 cal.methods = 1, 此时 scf.mixType参数会默认为Kerker
run程序运行
准备好输入文件 ioband.in 和 structure.as 上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW ioband.in 。
analysis计算结果分析
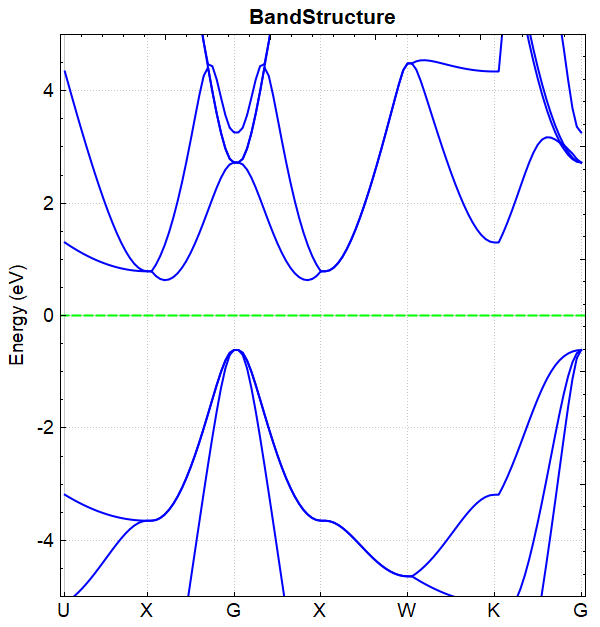
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 scf.h5 等输出文件。处理 scf.h5 的方法同(见2.3节)能带计算的方法,处理得到的能带图效果应如下所示:
修改杂化泛函Alpha系数
2.10.1章节展示的杂化泛函方法为HSE06,其对应的杂化泛函系数 sys.hybridAlpha = 0.25 , 调整 sys.hybridAlpha 参数作以下两次计算:
scf.in 和 band.in 中修改参数:
sys.hybridAlpha = 0.20scf.in 和 band.in 中修改参数:
sys.hybridAlpha = 0.30
得到如下能带对比图:
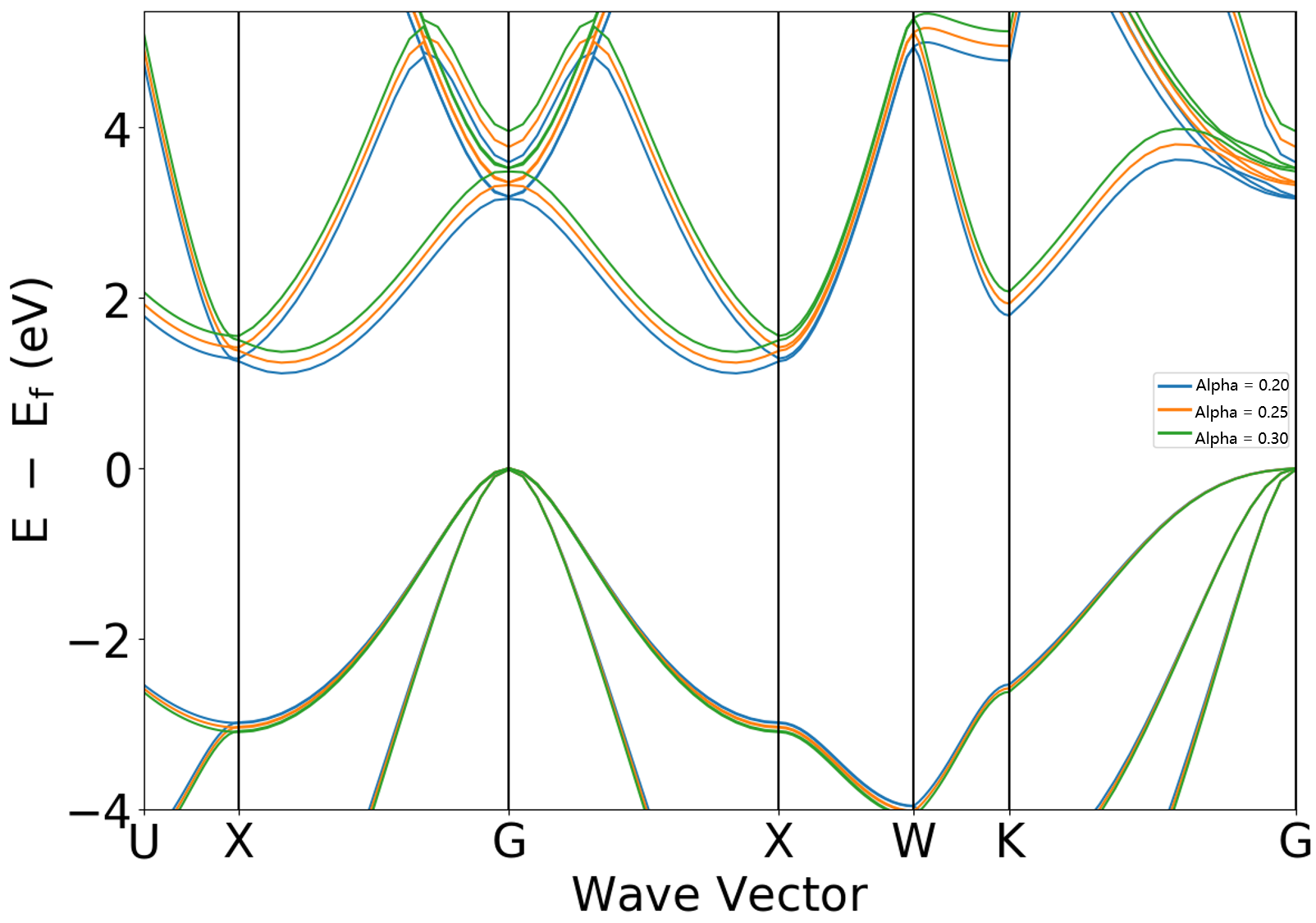
分析该图可得通过加大 sys.hybridAlpha 系数可以使能带带隙进一步增大。
从 DS-PAW.log 文件中可读取当 sys.hybridAlpha 分别取值 0.20 、 0.25 、 0.30 时,对应带隙值分别为 1.1146 、1.2394 、 1.3665 eV。
2.11 vdw范德瓦尔斯修正计算
本节将以石墨体系的结构弛豫为例,介绍在DS-PAW中如何正确的设置范德瓦尔斯修正,并将设置范德瓦尔斯修正与不设置该参数的结果进行对比分析。
graphite石墨结构弛豫输入文件
在对石墨进行弛豫时,可选取半经验方法对范德瓦尔斯力进行修正,也可采取泛函修正的方法,下面介绍两种方法下对应的参数设置。
半经验修正
输入文件包含参数文件 relax.in 和结构文件 structure.as , relax.in 如下:
1# task type
2task = relax
3#system related
4sys.structure = structure.as
5sys.symmetry = true
6sys.functional = PBE
7sys.spin = none
8#scf related
9cal.methods = 1
10cal.smearing = 1
11cal.ksamping = G
12cal.kpoints = [21, 21, 7]
13cal.cutoff = 600
14scf.convergence = 1.0e-05
15#relax related
16relax.max = 60
17relax.freedom = all
18relax.convergence = 0.01
19relax.methods = CG
20#vdw related
21corr.VDW = true
22corr.VDWType = D3G
relax.in 输入参数介绍:
在范德瓦尔斯修正计算中可以尽量保留sys.和cal.的参数到 relax.in 中,之后设置范德瓦尔斯修正计算特有的参数即可:
corr.VDW: 控制半经验方法范德瓦尔斯修正的开关,true表示已打开;corr.VDWType: 设置范德瓦尔斯修正的类型,D3G表示DFT-D3 of Grimme方法;
structure.as 文件参考如下:
1Total number of atoms
24
3Lattice
42.46729136 0.00000000 0.00000000
5-1.23364568 2.13673699 0.00000000
60.00000000 0.00000000 7.80307245
7Cartesian
8C 0.00000000 0.00000000 1.95076811
9C 0.00000000 0.00000000 5.85230434
10C 0.00000000 1.42449201 1.95076811
11C 1.23364689 0.71224492 5.85230434
备注
使用半经验方法对范德瓦尔斯力进行修正时,可选取不同类型的交换关联泛函,sys.functional 的可选值有 PBE/REVPBE/RPBE/PBESOL
DS-PAW支持使用半经验方法对范德瓦尔斯力进行修正的同时开启杂化泛函计算。
泛函修正
泛函修正对应的输入文件 relax.in 可如下所示:
1# task type
2task = relax
3#system related
4sys.structure = structure.as
5sys.symmetry = true
6sys.spin = none
7#scf related
8cal.methods = 1
9cal.smearing = 1
10cal.ksamping = G
11cal.kpoints = [21, 21, 7]
12cal.cutoff = 600
13scf.convergence = 1.0e-05
14#relax related
15relax.max = 60
16relax.freedom = all
17relax.convergence = 0.01
18relax.methods = CG
19#vdw related
20sys.functional = vdw-optPBE
relax.in 输入参数介绍:
在范德瓦尔斯修正计算中可以尽量保留sys.和cal.的参数到 relax.in 中,之后设置范德瓦尔斯修正计算特有的参数即可:
sys.functional: 控制泛函的类型,当选择包含范德瓦尔斯修正的泛函时,设置vdw-系列的泛函参数即可,此例选取 vdw-optPBE 泛函,支持的泛函类型见 参数说明 部分。
备注
从原理出发,范德瓦尔斯有两种不同的修正方式,分别对应参数 corr.VDW = true (半经验修正) 和 sys.functional = vdw-… (泛函修正)。
run程序运行
以半经验修正为例,准备好输入文件之后,将 relax.in 和 structure.as 文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW relax.in 。
analysis计算结果分析
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 relax.h5 、 latestStructure.as 等输出文件。(为作对比另添加一组不考虑范德瓦尔斯的计算)
将 latestStructure.as 拖入Device Studio查看结构,可得弛豫结束后晶胞常数如下表所示,通过对比可发现添加范德瓦尔斯修正进行结构弛豫所得晶胞向量 c 的值与实验报道结果[1] 更接近。
Procedure
a (Å)
c (Å)
vdw-D3G this work
2.463
6.954
PBE this work
2.464
7.914
Experiment
2.462
6.707
2.12 optical光学性质计算
光学计算有两种方式可完成,task=optical 的两步法及 task=scf 的一步法。本节将以Si体系为例,介绍在DS-PAW中如何进行光学性质的计算,并对一系列光学性质的物理量进行作图分析。
\(Si\) 光学性质计算输入文件
task = optical 两步算
输入文件包含参数文件 scf.in 、 optical.in 和结构文件 structure.as , scf.in 设置与自洽计算一致, optical.in 设置如下:
optical.in 如下:
1# task type
2task = optical
3#system related
4sys.structure = structure.as
5sys.symmetry = true
6sys.functional = PBE
7sys.spin = none
8#scf related
9cal.methods = 1
10cal.smearing = 1
11cal.ksamping = G
12cal.kpoints = [12, 12, 12]
13cal.cutoffFactor = 1.5
14cal.iniCharge = ./rho.bin
15
16#optical related
17optical.grid = 2000
18optical.sigma = 0.05
19optical.smearing = 1
在光学计算中可以尽量保留 sys. 和 cal. 的参数到 optical.in 中,之后设置光学计算特有的参数即可:
task: 设置计算类型,本次计算为task = optical:光学计算;cal.iniCharge: 设置读取电荷密度文件的路径,支持绝对路径及相对路径,这里./表示当前路径下的 rho.bin ;optical.grid: 表示DS-PAW计算光学性质时在能量区内的网格点数,此例为2000;optical.sigma: 决定使用optical.smearing决定的展开算法时的展宽宽度,此例为0.05;optical.smearing: 决定在optical计算时对能量展宽时的展宽算法,此例为1。
task = scf 一步算
输入文件包含参数文件 scf.in 和结构文件 structure.as , scf.in 设置如下:
1# task type
2task = scf
3#system related
4sys.structure = structure.as
5sys.symmetry = true
6sys.functional = PBE
7sys.spin = none
8#scf related
9cal.methods = 1
10cal.smearing = 1
11cal.ksamping = G
12cal.kpoints = [12, 12, 12]
13cal.cutoffFactor = 1.5
14#optical related
15io.optical = true
scf.in 输入参数介绍:
在光学性质计算中可以尽量保留sys.和cal.的参数到 scf.in 中,之后设置光学性质计算特有的参数即可:
io.optical: 控制光学性质计算的开关,当io.optical=true时,对体系进行光学性质的计算;
structure.as 文件同自洽计算。(见2.2节)
run程序运行
以两步算为例,准备好输入文件之后,将 scf.in 、 optical.in 和 structure.as 文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW scf.in 、 optical.in 。
analysis计算结果分析
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 scf.h5 、 optical.h5 等输出文件。
DS-PAW.log :DS-PAW光学性质计算之后得到的日志文件;
optical.h5 :光学性质计算完成之后的 h5 数据文件,注意h5文件的名称与task类型严格一致。h5文件的数据结构详见 输出文件格式说明 部分;
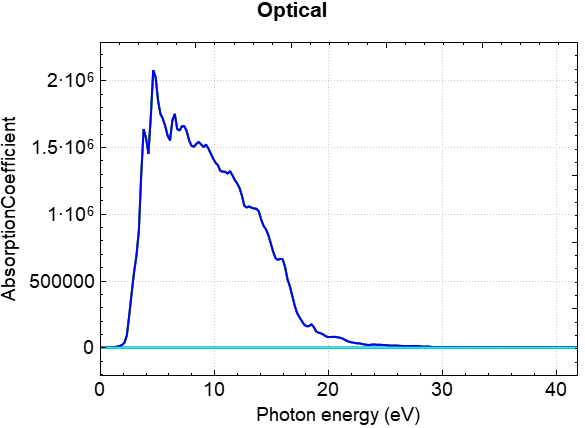
可使用 python 对 optical.h5 或者一步算的结果 scf.h5 进行数据处理,具体操作见 辅助工具使用教程 部分。处理可得介电常数实部、介电常数虚部、吸光系数、消光系数、光电导率、反射率、折射率、能量损失随能量的变化的曲线,以吸光系数曲线为例,得到的曲线效果图应如下所示:
2.13 frequency频率计算
本节将以 \(CO\) 分子为例,介绍在DS-PAW中如何进行频率计算。
\(CO\) 频率计算输入文件
输入文件包含参数文件 frequency.in 和结构文件 structure.as , frequency.in 如下:
1# task type
2task = frequency
3#system related
4sys.structure = structure.as
5sys.symmetry = true
6sys.functional = PBE
7sys.spin = none
8#scf related
9cal.methods = 2
10cal.smearing = 1
11cal.ksamping = MP
12cal.kpoints = [9, 9, 9]
13cal.cutoffFactor = 1.5
14scf.convergence = 1.0e-6
15#frequency related
16frequency.dispOrder = 1
17frequency.dispRange = 0.02
18#outputs
19io.charge = false
20io.wave = false
frequency.in 输入参数介绍:
在频率计算中可以尽量保留sys.和cal.的参数到 frequency.in 中,之后设置频率计算特有的参数即可:
task: 设置计算类型,本次计算为frequency频率计算;frequency.dispOrder: 设置频率计算时原子振动的方式。1对应中心差分法,即2种原子振动方式:每个笛卡尔方向上原子的位移为±frequency.dispRange;2对应4种原子振动方式:每个笛卡尔方向上原子的位移为±frequency.dispRange和±2*frequency.dispRange;frequency.dispRange: 设置频率计算时的原子位移大小;
structure.as 文件参考如下:
1Total number of atoms
22
3Lattice
48.0 0.0 0.0
50.0 8.0 0.0
60.0 0.0 8.0
7Cartesian Fix_x Fix_y Fix_z
8O 0 0 0 T T F
9C 0 0 1.143 T T F
备注
频率计算时应提高自洽计算的收敛精度,建议设置为1.0e-6以上。
由于固定了C,O原子在x,y方向的自由度,故两原子只在z方向上可动。
run程序运行
准备好输入文件之后,将 frequency.in 和 structure.as 文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW frequency.in 。
analysis计算结果分析
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 frequency.h5 、 frequency.txt 等输出文件。
DS-PAW.log :DS-PAW频率计算之后得到的日志文件;
frequency.h5 :频率计算完成之后的h5数据文件,此时频率数据被保存在该文件中,具体的数据结构详见 输出文件格式说明 部分。
frequency.txt :频率计算完成之后的 txt 文本文件,该文件写入频率相关数据,与 frequency.h5 文件数据一致,便于用户快速获取信息。
从 frequency.txt 中可获取以下数据:
Frequency |
THz |
2PiTHz |
cm-1 |
meV |
1 f |
63.844168 |
401.144726 |
2129.612084 |
264.038342 |
2 f/i |
0.051335 |
0.322546 |
1.712346 |
0.212304 |
CO只在z方向上两个原子可动,因此只有两个频率,通过上表可以看到一个振动模式的频率约为 63.8 THz,还有一个在 0 附近的虚频,一般情况虚频小于2THz基本可以忽略不计。
2.14 elastic弹性常数计算
本节将以Si体系为例,介绍在DS-PAW中如何进行弹性计算。
\(Si\) 弹性常数计算输入文件
输入文件包含参数文件 elastic.in 和结构文件 structure.as , elastic.in 如下:
1# task type
2task = elastic
3#system related
4sys.structure = structure.as
5sys.symmetry = true
6sys.functional = PBE
7sys.spin = none
8#scf related
9cal.methods = 1
10cal.smearing = 1
11cal.ksamping = G
12cal.kpoints = [5, 5, 5]
13cal.cutoffFactor = 1.5
14scf.convergence = 1.0e-6
15#frequency related
16elastic.dispOrder = 1
17elastic.dispRange = 0.01
18#outputs
19io.charge = false
20io.wave = false
elastic.in 输入参数介绍:
在弹性计算中可以尽量保留sys.和cal.的参数到 elastic.in 中,之后设置弹性计算特有的参数即可:
task: 设置计算类型,本次计算为elastic弹性计算;elastic.dispOrder: 设置弹性计算时原子振动的方式, 1对应中心差分法;elastic.dispRange: 设置弹性计算时原子位移的大小;
structure.as 文件参考如下:
1Total number of atoms
28
3Lattice
45.43070000 0.00000000 0.00000000
50.00000000 5.43070000 0.00000000
60.00000000 0.00000000 5.43070000
7Cartesian
8Si 0.67883750 0.67883750 0.67883750
9Si 3.39418750 3.39418750 0.67883750
10Si 3.39418750 0.67883750 3.39418750
11Si 0.67883750 3.39418750 3.39418750
12Si 2.03651250 2.03651250 2.03651250
13Si 4.75186250 4.75186250 2.03651250
14Si 4.75186250 2.03651250 4.75186250
15Si 2.03651250 4.75186250 4.75186250
备注
弹性计算时应提高自洽计算的收敛精度,建议设置在1.0e-6以上。
弹性计算不支持固定原子。
run程序运行
准备好输入文件之后,将 elastic.in 和 structure.as 文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW elastic.in 。
analysis计算结果分析
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 elastic.h5 、 elastic.txt 这3个文件。
DS-PAW.log :DS-PAW弹性计算之后得到的日志文件;
elastic.h5 : 弹性计算完成之后的 h5 数据文件,此时弹性模量被保存在 elastic.h5 中,具体的数据结构详见 输出文件格式说明 部分;
elastic.txt : 弹性计算完成之后的 txt 文本文件,该文件写入弹性相关数据,与 elastic.h5 文件数据一致,便于用户快速获取信息。
从 elastic.txt 文件可得如下弹性常数矩阵:
刚性弹性矩阵:
158.7644
62.9858
62.9858
0.0000
-0.0000
0.0000
62.9858
158.7644
62.9858
0.0000
0.0000
0.0000
62.9858
62.9858
158.7644
-0.0000
0.0000
0.0000
0.0000
0.0000
-0.0000
75.8807
-0.0000
0.0000
-0.0000
0.0000
0.0000
-0.0000
75.8807
-0.0000
0.0000
0.0000
0.0000
0.0000
-0.0000
75.8807
柔性弹性矩阵:
0.0081
-0.0023
-0.0023
-0.0000
0.0000
-0.0000
-0.0023
0.0081
-0.0023
-0.0000
-0.0000
0.0000
-0.0023
-0.0023
0.0081
0.0000
-0.0000
0.0000
-0.0000
-0.0000
0.0000
0.0132
0.0000
-0.0000
0.0000
-0.0000
-0.0000
0.0000
0.0132
0.0000
-0.0000
0.0000
0.0000
-0.0000
0.0000
0.0132
体积模量、剪切模量、杨氏模量和泊松比:
Properties
Vogit
Reuss
Hill
BulkModulus(GPa)
94.9120
94.9120
94.9120
ShearModulus(GPa)
64.6841
61.5016
63.0929
YoungModulus(GPa)
158.1297
151.7315
154.9452
PoissonRatio
0.2223
0.2336
0.2279
Si体系为Cubic晶系,该晶系的独立矩阵元有三个:C11 , C12 , C44 ,分别对应表中的 158.7644、62.9858、75.8807。
2.15 neb过渡态计算
本节将以H在Pt(100)表面扩散为例,介绍在DS-PAW中如何进行过渡态计算(Cl-NEB),并对结果进行作图分析。
\(Pt\) 过渡态计算输入文件
输入文件包含参数文件 neb.in 和多个结构文件 structureNo.as , neb.in 文件如下:
1task = neb
2
3sys.structure = structure.as
4sys.functional = PBE
5sys.spin = none
6sys.symmetry = true
7
8cal.ksamping = G
9cal.kpoints = [3,3,1]
10cal.cutoffFactor = 1.0
11cal.smearing = 1
12cal.sigma = 0.05
13
14neb.freedom = atom
15neb.springK = 5
16neb.images = 3
17neb.iniFin = true
18neb.method = LBFGS
19neb.convergence = 0.03
20neb.stepRange = 0.1
21neb.max = 60
22
23io.wave = false
24io.charge = false
neb.in 输入参数介绍:
在过渡态计算中可以尽量保留sys.和cal.的参数到 neb.in 中,之后设置过渡态计算特有的参数即可:
task:设置计算类型,本次计算为neb过渡态计算;neb.stepRange: 设置过渡态计算中结构弛豫的步长;neb.max: 设置过渡态计算中结构弛豫的最大步数;neb.iniFin: 控制过渡态计算中初态结构和末态结构是否进行自洽计算,true表示进行自洽计算;neb.springK: 设置过渡态计算中弹簧系数K;neb.images: 设置过渡态计算中的中间结构的数目;neb.method: 设置过渡态计算中使用的算法;neb.convergence: 设置过渡态计算中受力的收敛标准;
structure.as 需提供多个,初态结构 structure00.as 参考如下
1Total number of atoms
213
3Lattice
45.60580000 0.00000000 0.00000000
50.00000000 5.60580000 0.00000000
60.00000000 0.00000000 16.81740000
7Cartesian Fix_x Fix_y Fix_z
8H 2.80881670 4.20393628 6.94088012 F F F
9Pt 1.40145000 1.40145000 1.98192999 T T T
10Pt 4.20434996 1.40145000 1.98192999 T T T
11Pt 1.40145000 4.20434996 1.98192999 T T T
12Pt 4.20434996 4.20434996 1.98192999 T T T
13Pt 0.00272621 0.00056545 3.91746017 F F F
14Pt 0.00271751 2.80233938 3.91708172 F F F
15Pt 2.80568712 -0.00141176 3.91894328 F F F
16Pt 2.80548220 2.80426217 3.91792247 F F F
17Pt 1.39865124 1.40124680 5.84694340 F F F
18Pt 4.21951864 1.40156999 5.84719575 F F F
19Pt 1.38647954 4.20437926 5.89984296 F F F
20Pt 4.23154392 4.20414605 5.89983612 F F F
末态结构 structure04.as 参考如下
1Total number of atoms
213
3Lattice
45.60580000 0.00000000 0.00000000
50.00000000 5.60580000 0.00000000
60.00000000 0.00000000 16.81740000
7Cartesian Fix_x Fix_y Fix_z
8H 1.52157824 2.80289997 6.91583941 F F F
9Pt 1.40145000 1.40145000 1.98192999 T T T
10Pt 4.20434997 1.40145000 1.98192999 T T T
11Pt 1.40145000 4.20434997 1.98192999 T T T
12Pt 4.20434997 4.20434997 1.98192999 T T T
13Pt 0.02556963 0.00000000 3.90765450 F F F
14Pt 0.02708862 2.80290000 3.91082177 F F F
15Pt 2.83159105 0.00000000 3.91547525 F F F
16Pt 2.82981856 2.80290000 3.90913282 F F F
17Pt 1.45998966 1.38039927 5.88134827 F F F
18Pt 4.25691060 1.38811299 5.84551487 F F F
19Pt 1.45998966 4.22540069 5.88134827 F F F
20Pt 4.25691060 4.21768697 5.84551487 F F F
备注
neb计算时初态末态需先进行结构弛豫。
中间结构的生成可调用“辅助工具使用教程-过渡态部分”的neb_interpolate_structures.py脚本,完成插值可调用neb_visualize.py脚本对插值结构进行预览,可调用calc_dist.py脚本查看image之间的距离是否合理。
过渡态计算时的结构文件structureNo.as需存放在命名为No的文件夹中,文件夹序号与结构文件的序号需一致。文件夹外放置一个neb.in文件即可,在neb.in所在目录执行DS-PAW程序。
过渡态计算执行程序时调用的核数设置为images的整数倍。
run程序运行
准备好输入文件之后,将 neb.in 文件和包含 structureNo.as 文件的多个文件夹文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW neb.in 。
analysis计算结果分析
根据上述的输入文件,计算完成之后:
>> 初态和末态结构所在文件夹会生成自洽计算所得的 DS-PAW.log 、latestStructure00.as 、 scf.h5 等输出文件;
>> 中间结构 structureNo.as 所在文件夹 No (参与过渡态计算的中间结构所在文件夹,中间结构的个数由 neb.images 参数决定)会生成结构优化所得的 nebNo.h5 、 latestStructureNo.as 等输出文件;
>> 最外层目录将会生成 DS-PAW.log 、 neb.h5 这2个文件,其中 neb.h5 为 No 文件夹下各 nebNo.h5 文件的信息汇总。
DS-PAW.log :DS-PAW过渡态计算之后得到的日志文件;
neb.h5 :过渡态计算完成之后的 h5 数据文件;此时反应坐标及能量变化等数据被保存在 neb.h5 中,具体的数据结构详见 输出文件格式说明 部分;
可使用 python 脚本 8neb_check_results.py 对neb计算的结果进行分析,需在完整的neb计算目录下执行分析脚本,具体操作见 辅助工具使用教程 部分。
处理可得到NEB计算各构型的能量和受力表格:
Image |
Force (eV/Å) |
Reaction coordinate (Å) |
Energy (eV) |
Delta energy (eV) |
00 |
0.1803 |
0.0000 |
-39637.0984 |
0.0000 |
01 |
0.0263 |
0.5428 |
-39637.0186 |
0.0798 |
02 |
0.0248 |
1.0868 |
-39636.8801 |
0.2183 |
03 |
0.2344 |
1.5884 |
-39636.9984 |
0.1000 |
04 |
0.0141 |
2.0892 |
-39637.0900 |
0.0084 |
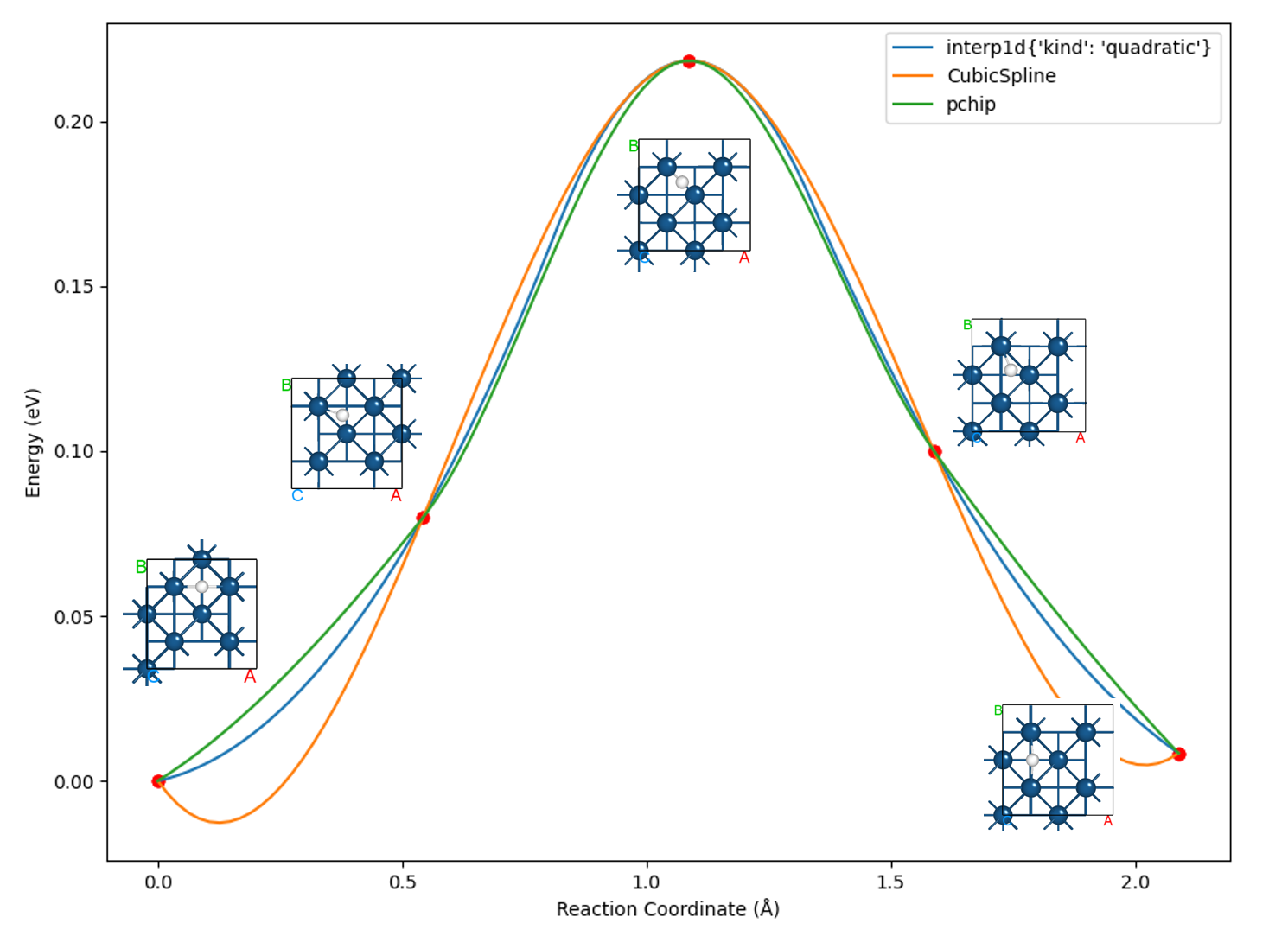
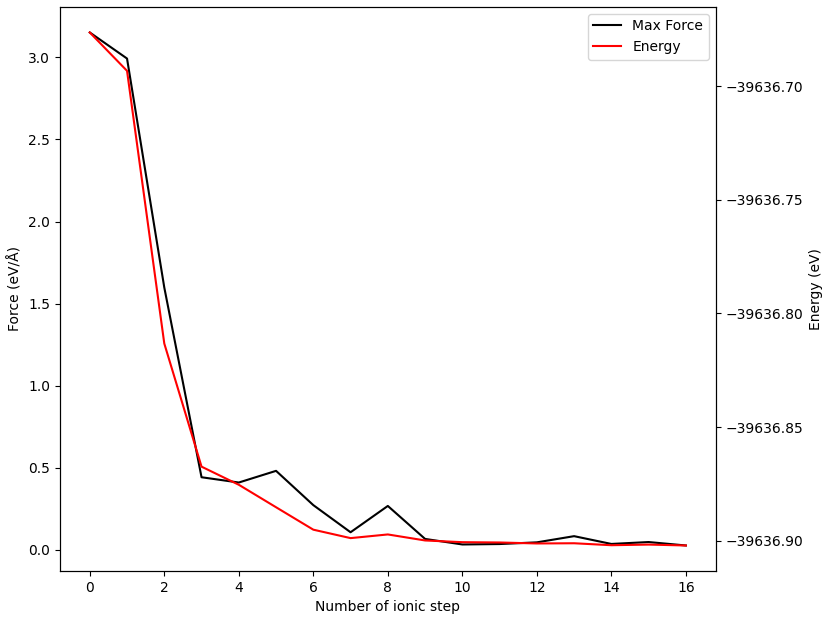
另可使用 python 脚本 neb_movie.py 分析过渡态搜寻中的轨迹变化,生成的 neb_movie.json 文件可用 Device Studio 打开,截取一帧如下所示:
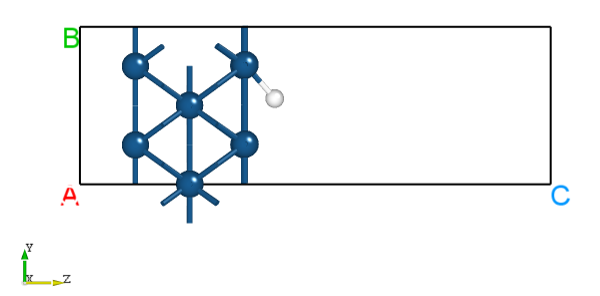
2.16 phonon声子谱计算
本节介绍DS-PAW程序如何进行声子计算及声子能带和声子态密度计算。DS-PAW支持两种声子谱计算的方法:fd有限位移法和dfpt密度泛函微扰理论方法。本节以单个MgO体系为例,介绍如何用两种方法计算声子能带和态密度,并对声子能带和态密度作图进行分析。
\(MgO\) 声子谱能带计算输入文件
输入文件包含参数文件 phonon.in 和结构文件 structure.as , phonon.in 如下:
1task = phonon
2
3sys.structure = structure.as
4sys.functional = PBE
5sys.spin = none
6
7cal.methods = 1
8cal.smearing = 1
9sys.symmetry = true
10scf.convergence = 1.0e-07
11cal.ksamping = G
12cal.kpoints = [3,3,3]
13cal.sigma = 0.25
14
15phonon.type = bandDos
16phonon.structureSize = [2,2,2]
17phonon.primitiveUVW = [0.0, 0.5, 0.5, 0.5, 0.0, 0.5, 0.5, 0.5, 0.0]
18phonon.method = dfpt
19phonon.qpoints = [41,41,41]
20phonon.dosRange = [0,20]
21phonon.qpointsLabel = [G,X,W,G,M]
22phonon.qpointsCoord = [0.0, 0.0, 0.0, 0.5, 0.0, 0.0, 0.5, 0.5, 0.0, 0.0, 0.0, 0.0, 0.5, 0.5, 0.5]
23phonon.qpointsNumber = 51
24
25io.charge = false
26io.wave = false
phonon.in 输入参数介绍:
在声子计算中可以尽量保留sys.和cal.的参数到 phonon.in 中,之后设置声子计算特有的参数即可:
task: 设置计算类型,本次计算为phonon声子计算;phonon.type: 设置声子计算的类型,bandDos对应计算声子能带和态密度;phonon.structureSize: 设置声子计算时超胞的大小;phonon.primitiveUVW: 设置声子能带计算时原胞UVW的系数;phonon.method: 设置声子计算的方法,dfpt为密度泛函微扰理论方法;phonon.qpoints: 设置声子计算q空间网格取样为41*41*41;phonon.dosRange: 设置声子态密度计算的能量区间为[0,20];phonon.qpointsLabel: 设置声子能带计算时高对称点标签;phonon.qpointsCoord: 设置声子能带计算时高对称点坐标;phonon.qpointsNumber: 设置声子能带相邻两个高对称点的间隔;
structure.as 文件参考如下:
1Total number of atoms
28
3Lattice
4 4.2555564654942897 0.0000000000000000 0.0000000000000000
5 0.0000000000000000 4.2555564654942888 0.0000000000000000
6 0.0000000000000000 0.0000000000000000 4.2555564654942897
7Direct
8Mg 0.0000000000000000 0.0000000000000000 0.0000000000000000
9Mg 0.0000000000000000 0.5000000000000000 0.5000000000000000
10Mg 0.5000000000000000 0.0000000000000000 0.5000000000000000
11Mg 0.5000000000000000 0.5000000000000000 0.0000000000000000
12O 0.5000000000000000 0.5000000000000000 0.5000000000000000
13O 0.5000000000000000 0.0000000000000000 0.0000000000000000
14O 0.0000000000000000 0.5000000000000000 0.0000000000000000
15O 0.0000000000000000 0.0000000000000000 0.5000000000000000
备注
声子计算时应提高自洽计算的收敛精度,建议设置在1.0e-7以上。
声子计算时若打开对称性,建议适当提高对称性判断的精度,参数sys.symmetryAccuracy可设置为1.0e-6或更小,助于得到准确的计算结果。
phonon.iniPhonon可指定路径读取声子计算(phonon.type = phonon)得到的phonon.h5文件,从而直接进行能带与态密度的计算。
phonon.type控制计算声子的类型,phonon对应计算声子,band对应计算声子能带,dos对应计算声子态密度,bandDos对应同时计算声子能带和态密度。当phonon.type = band/dos/bandDos且phonon.iniPhonon未指定文件路径时,程序先自动完成phonon.type = phonon的声子计算,然后根据任务计算能带或态密度。
run程序运行
准备好输入文件之后,将 phonon.in 和 structure.as 文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW phonon.in 。
analysis计算结果分析
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 phonon.h5 、 dfpt.json 和 dfpt.as 等输出文件。
DS-PAW.log :DS-PAW声子计算之后得到的日志文件;
dfpt.as :声子计算时超胞的结构文件,计算声子时读取该文件信息。
dfpt.json :声子计算时的参数文件,该文件与 phonon.in 文件信息一致,计算声子时读取该文件信息。
phonon.h5 :声子计算完成之后的 h5 数据文件;此时声子能带数据被保存在 phonon.h5 中,具体的数据结构详见 输出文件格式说明 部分;
可使用 python 脚本对 phonon.h5 进行数据处理,处理得到的声子能带和态密度效果图应如下(a)、(b)所示:
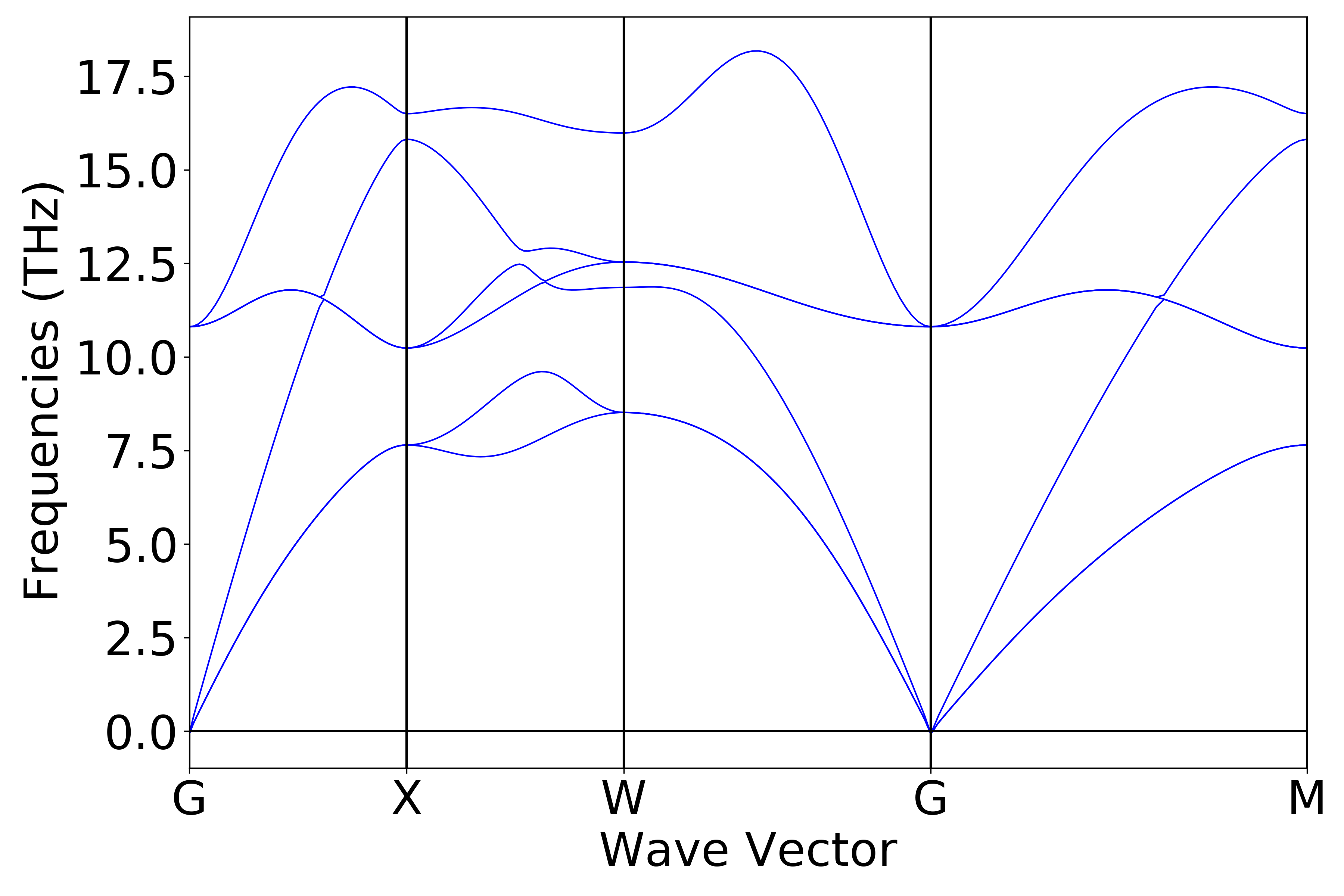
(a)
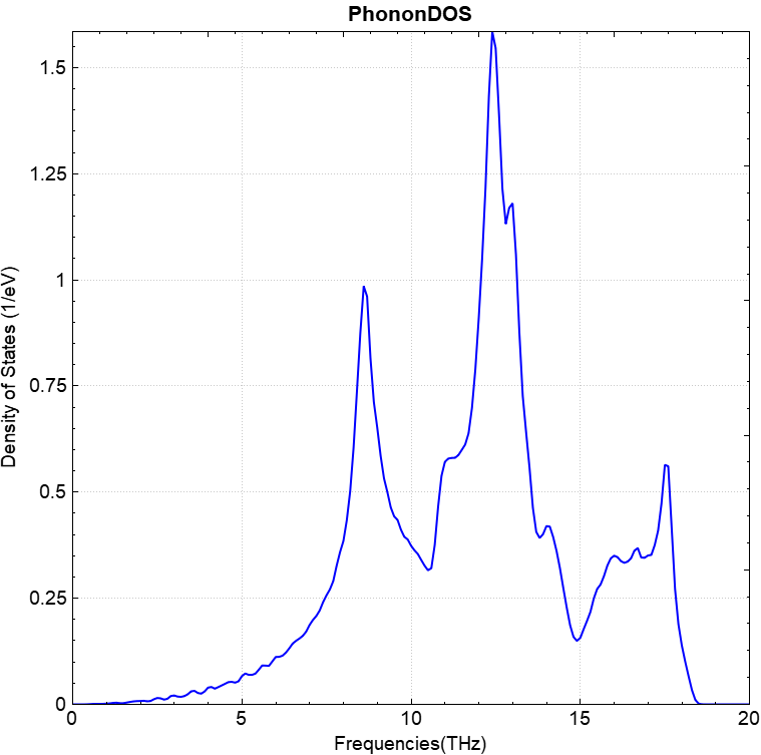
(b)
nac计算结果分析
上节展示的为不考虑长程相互作用的声子能带计算,若打开 non-analytical term correction (nac) 进行声子计算,在上节所示的 phonon.in 文件中添加以下两个参数即可:
1phonon.dfptEpsilon=true
2phonon.nac = true
得到的声子能带效果图应如下(c)所示:
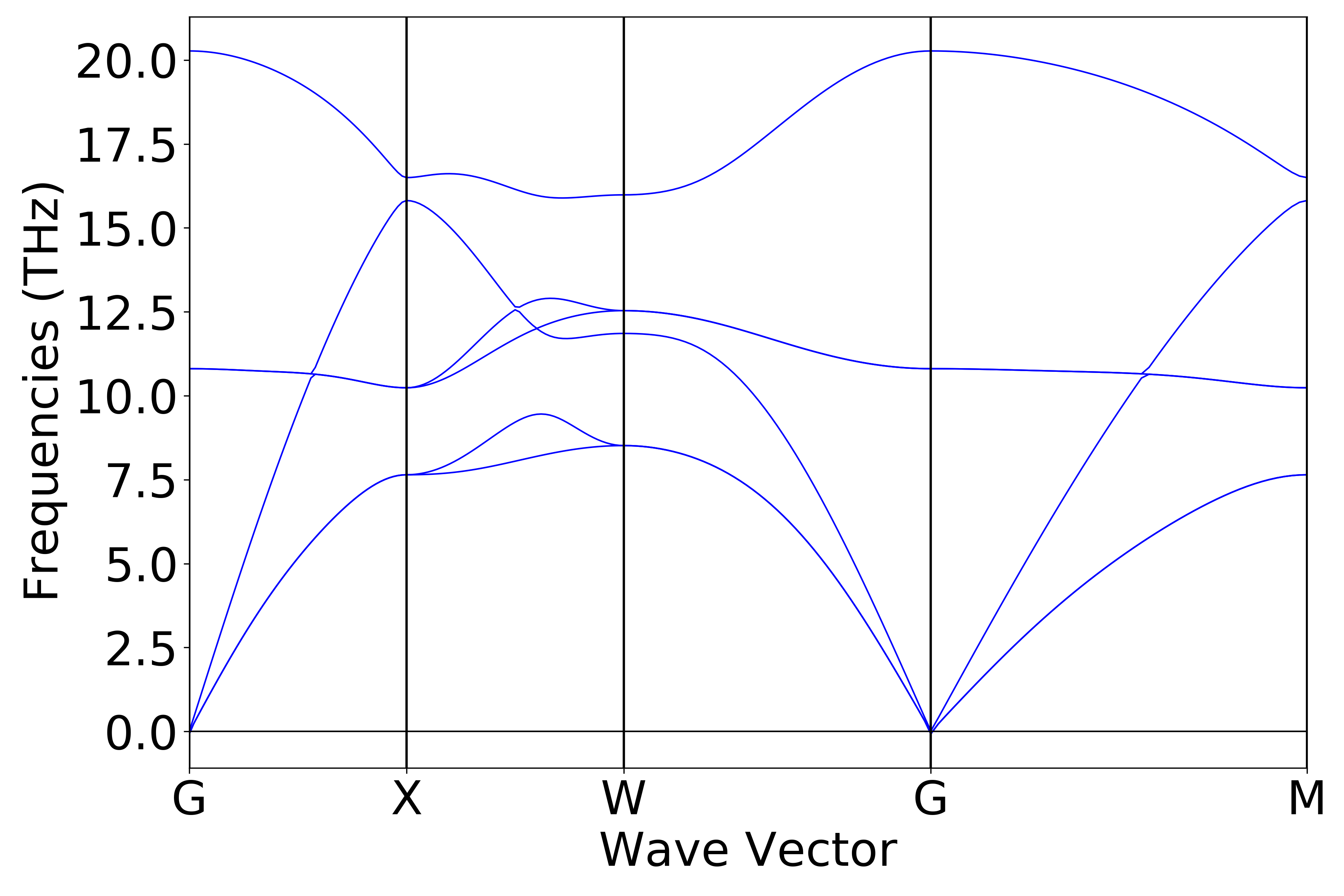
(c)
fdphonon有限位移法计算声子
有限位移法(fd) 计算的输入文件如下所示,将参数 phonon.method = dfpt 修改为 phonon.method = fd 即可,需要注意的是,fd方法计算得到输出文件与dfpt方法不同。
1task = phonon
2
3sys.structure = structure.as
4sys.functional = PBE
5sys.spin = none
6
7cal.methods = 1
8cal.smearing = 1
9sys.symmetry = true
10scf.convergence = 1.0e-07
11cal.ksamping = G
12cal.kpoints = [3,3,3]
13cal.sigma = 0.25
14
15phonon.type = bandDos
16phonon.structureSize = [2,2,2]
17phonon.primitiveUVW = [0.0, 0.5, 0.5, 0.5, 0.0, 0.5, 0.5, 0.5, 0.0]
18phonon.method = fd
19phonon.qpoints = [41,41,41]
20phonon.qpointsLabel = [G,X,W,G,M]
21phonon.qpointsCoord = [0.0, 0.0, 0.0, 0.5, 0.0, 0.0, 0.5, 0.5, 0.0, 0.0, 0.0, 0.0, 0.5, 0.5, 0.5]
22phonon.qpointsNumber = 51
23
24io.charge = false
25io.wave = false
以MgO体系为例, phonon.structureSize 设置为 [2,2,2] ,fd法计算完成之后将会得到 DS-PAW.log 、 phonon.h5 两个文件 和 001 、 002 文件夹。 001 文件夹下存在 input.json 和 disp-001.as 文件,002 文件夹下存在 input.json 和 disp-002.as 文件,子文件夹中的两个文件等同于写入输入参数的 in 文件和结构参数的 as 文件,
生成文件夹(001 002…)的个数取决于体系的对称性。
用 python 脚本处理有限位移方法计算得到的 phonon.h5 文件,得到能带图及态密度图同dfpt方法计算得到的图(a)与(b)一致。
备注
介电常数的计算只在phonon.method = dfpt时才能完成
phonon.nac的开关只在phonon.method = dfpt且phonon.dfptEpsilon=true时生效
2.17 soc自旋轨道耦合计算
本节介绍DS-PAW如何进行自旋轨道耦合计算。以 \(Bi_{2}Se_{3}\) 体系为例,使用两步法进行能带计算并对能带进行作图分析。
\(Bi_{2}Se_{3}\) 自旋轨道耦合计算输入文件
首先进行自洽计算:输入文件包含参数文件 soi.in 和结构文件 structure.as , soi.in 如下:
1# task type
2task = scf
3#system related
4sys.structure = structure.as
5sys.symmetry = false
6sys.functional = PBE
7#scf related
8cal.methods = 2
9cal.smearing = 1
10cal.ksamping = G
11cal.kpoints = [7, 7, 7]
12cal.cutoffFactor = 1.5
13#soi related
14sys.spin= non-collinear
15sys.soi = true
16#outputs
17io.charge = true
18io.wave = false
soi.in 输入参数介绍:
在自旋轨道耦合计算中可以尽量保留sys.和cal.的参数到 soi.in 中,之后设置自旋轨道耦合计算特有的参数即可:
sys.spin: 设置体系自旋类型,non-collinear 表示非线性自旋;sys.soi: 控制是否考虑自旋轨道耦合效应;该参数在sys.spin=non-collinear时生效;
structure.as 文件参考如下:
1Total number of atoms
25
3Lattice
4-2.069 -3.583614 0.000000
52.069 -3.583614 0.000000
60.000 2.389075 9.546667
7Direct
8Bi 0.3990 0.3990 0.6970
9Bi 0.6010 0.6010 0.3030
10Se 0.0000 0.0000 0.5000
11Se 0.2060 0.2060 0.1180
12Se 0.7940 0.7940 0.8820
能带计算的输入文件 soiband.in ,内容如下
1# task type
2task = band
3#system related
4sys.structure = structure.as
5sys.symmetry = true
6sys.functional = PBE
7#scf related
8cal.methods = 2
9cal.smearing = 1
10cal.ksamping = G
11cal.kpoints = [7, 7, 7]
12cal.cutoffFactor = 1.5
13#band related
14cal.iniCharge = ./rho.bin
15band.kpointsCoord = [0.00000000,0.00000000,0.00000000,0.00000000,0.00000000,0.50000000,0.50000000,0.50000000,0.00000000,0.00000000,0.00000000,0.00000000,0.50000000,0.00000000,0.00000000]
16band.kpointsLabel = [G,Z,F,G,L]
17band.kpointsNumber = [20,20,20,20]
18band.project = true
19#soi related
20sys.spin= non-collinear
21sys.soi = true
soiband.in 输入参数介绍:
在自旋轨道耦合能带计算中,保留自洽计算和自旋轨道耦合计算的参数到 soiband.in 中,之后设置能带计算的特有参数即可。
备注
初始磁矩的设置参考“应用案例-NiO体系的反铁磁计算”,在structure.as文件的第七行设置Mag标签即可。
run程序运行
准备好输入文件之后,将 soi.in 、 soiband.in 和 structure.as 文件上传到服务器上运行,按照结构弛豫中介绍的方法分别执行 DS-PAW soi.in 和 DS-PAW soiband.in 。
analysis计算结果分析
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 scf.h5 、 band.h5 等输出文件。
处理 band.h5 的方法同(见2.3节)能带计算的方法,得到的能带效果图应如下图(a)所示,另作不考虑自旋轨道耦合的计算,得到的能带效果图应如下(b)所示:
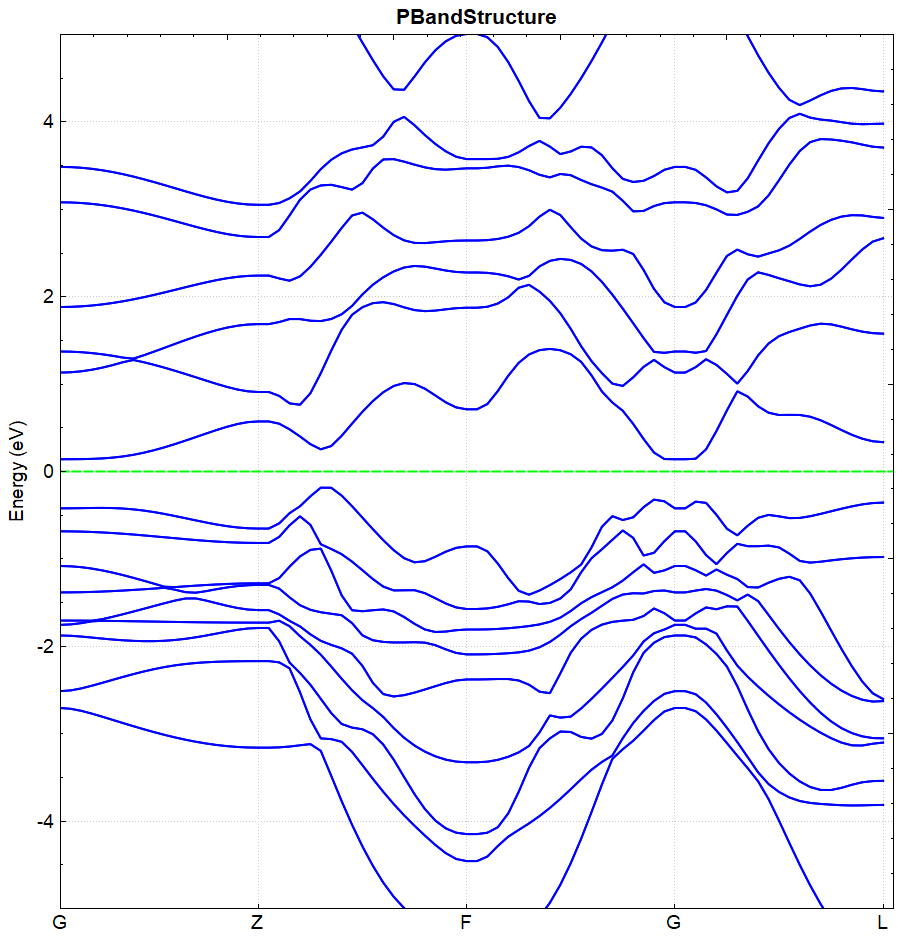
(a)
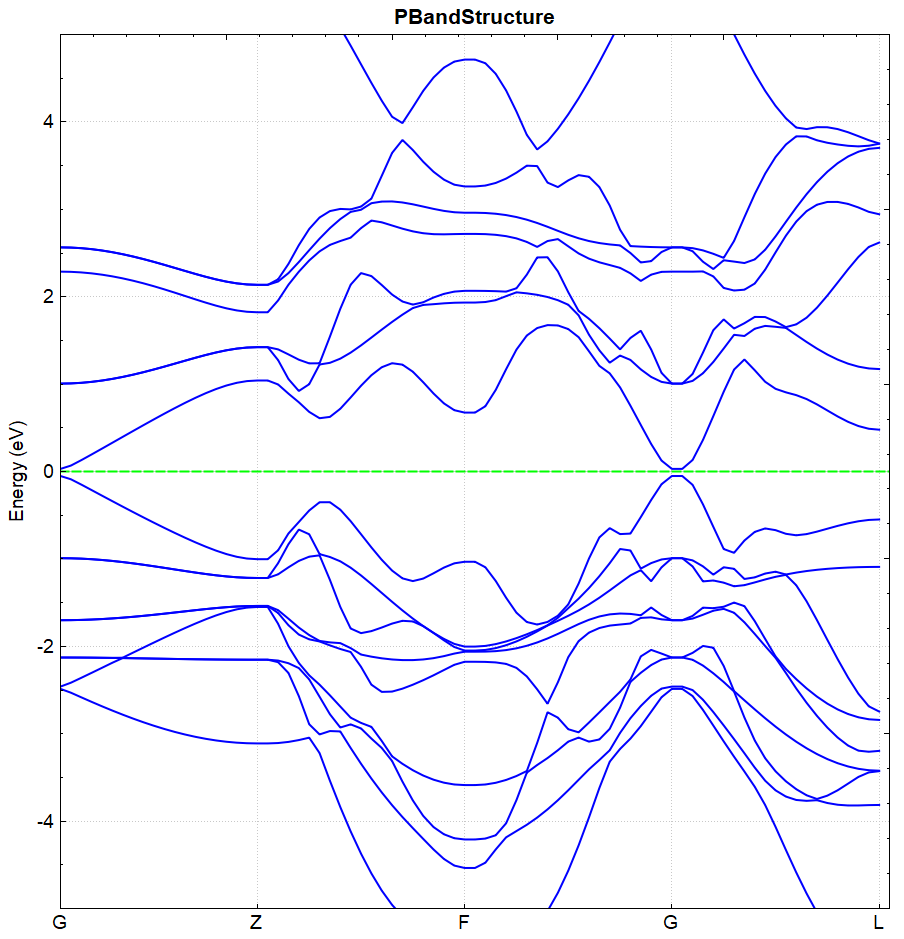
(b)
从 DS-PAW.log 可读取 BandGap 值,图(a)和图(b)的带隙值分别为 0.3251 eV 和 0.0814 eV ,可得结论:进行自旋轨道耦合计算可加大价带与导带之间的带隙。
2.18 aimd分子动力学模拟
本节将以水分子体系为例,介绍在DS-PAW中如何进行分子动力学模拟计算。
\(H_{2}O\) 分子动力学模拟输入文件
输入文件包含参数文件 aimd.in 和结构文件 structure.as , aimd.in 如下:
1#task type
2task = aimd
3
4#system related
5sys.structure = structure.as
6sys.symmetry = false
7sys.functional = PBE
8sys.spin = none
9
10#scf related
11cal.methods = 1
12cal.smearing = 1
13cal.ksamping = G
14cal.kpoints = [1, 1, 1]
15cal.sigma = 0.1
16
17#aimd related
18aimd.ensemble = NPT
19aimd.thermostat = langevin
20aimd.atomFCoeffElements = [H_1]
21aimd.atomFCoeffs = [1]
22aimd.latticeFCoeff = 1
23aimd.pressure = 100
24aimd.timeStep = 1
25aimd.totalSteps = 2000
26aimd.iniTemp = 2000
27
28#outputs
29io.charge = false
30io.wave = false
aimd.in 输入参数介绍:
在分子动力学模拟计算中可以尽量保留sys.和cal.的参数到 aimd.in 中,之后设置分子动力学模拟计算特有的参数即可:
task: 设置计算类型,本次计算为aimd分子动力学模拟计算;aimd.ensemble: 设置分子动力学模拟时选用的系综,此例系综设置为 NPT ;aimd.thermostat:设置分子动力学模拟时选用的恒温器或恒压器,此例选用 langevin 控温控压;aimd.atomFCoeffElements: 设置考虑为 langevin 原子的元素名称,此例将其中一个氢原子设置为 langevin 原子,将该氢原子重命名为 H_1 ;aimd.atomFCoeffs: 设置考虑为 langevin 原子对应的摩擦系数,单位 ps-1;aimd.latticeFCoeff: 设置 langevin 恒温器中晶胞摩擦系数的大小,单位 ps-1;aimd.pressure: 设置 NPT 模拟时体系的目标压强值,单位 kbar;aimd.timeStep: 设置分子动力学模拟时的时间步长,单位 fs;aimd.totalSteps: 设置分子动力学模拟的总步数;aimd.iniTemp: 设置分子动力学模拟时的初始温度,单位 K;
structure.as 文件参考如下:
1Total number of atoms
23
3Lattice
44.00000000 0.00000000 0.00000000
50.00000000 4.00000000 0.00000000
60.00000000 0.00000000 4.00000000
7Cartesian
8H 2.63934013 1.89542007 1.58223984
9H_1 1.36065987 2.11498988 2.45934006
10O 1.65002999 1.88501012 1.54065994
备注
元素重命名规则为 “原元素名 +下划线 + 自定义字段” 。
此例中设置第二个氢原子为 langevin 原子并将其重命名为 H_1, 在structure.as文件中需手动对该原子的元素名称进行修改。
计算体系中存在自定义元素名称 H_1,程序会自动查找 H_1 对应的 H 元素的赝势,用户无需额外准备新的赝势。
run程序运行
准备好输入文件之后,将 aimd.in 和 structure.as 文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW aimd.in 。
analysis计算结果分析
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 aimd.h5 、 latestStructure.as 等输出文件。
DS-PAW.log :DS-PAW分子动力学模拟计算得到的日志文件;
aimd.h5 : 分子动力学计算对应的 h5 输出文件;此时模拟时间内原子位置、体系能量和温度等数据被保存在 aimd.h5 中,具体的数据结构详见 输出文件格式说明 部分;
latestStructure.as : 分子动力学模拟计算所得的末态as结构文件,保存末态构型和速度信息;
可使用 python 脚本对 aimd.h5 文件进行数据处理,具体操作见 辅助工具使用教程 部分。在 NPT 系综下模拟 2000 步得到的 压力随时间 、 温度随时间 变化曲线效果图应如下所示:

备注
不同系综对应可选热浴范围:NVE系综可选andersen热浴、NVT系综可选andersen、noseHoover和langevin三种热浴,NPT系综可选langevin热浴、NPH系综可选langevin热浴;
如需模拟高温退火过程,设置 aimd.ensemble 为 SA,同时通过 aimd.iniTemp 和 aimd.finTemp 设置初始和末态温度即可;
参数 aimd.finTemp 只在模拟退火时生效,恒温系综如 NPT 和 NVT,末态温度等于初态温度。
当模拟体系中含 langevin 原子时,建议将 langevin 原子对应的赝势文件存放在计算目录,以避免程序找不到赝势文件报错 E3058。
2.19 efield外加电场计算
本节将以硅烯模型的能带计算为例,介绍在DS-PAW中如何进行外加电场计算,分析加电场前后带隙打开情况。
硅烯真空方向外加电场计算输入文件
输入文件包含参数文件 Efield.in 和结构文件 structure.as , Efield.in 如下:
1# task type
2task = scf
3#system related
4sys.structure = structure.as
5sys.symmetry = true
6sys.functional = PBE
7sys.spin = none
8
9#scf related
10cal.sigma = 0.1
11cal.cutoff = 520
12cal.ksamping = G
13cal.kpoints = [9, 9, 1]
14
15scf.convergence = 1e-5
16
17#outputs
18io.charge = false
19io.wave = false
20io.band = true
21
22corr.dipol=true
23corr.dipolDirection = c
24corr.dipolEfield = 0.2
25
26band.kpointsLabel = [G,M,K,G]
27band.kpointsCoord = [0.00000000,0.00000000,0.00000000,0.50000000,0.00000000,0.00000000,0.33333333,0.33333333,0.00000000,0.00000000,0.00000000,0.00000000]
28band.kpointsNumber = [100,100,100]
Efield.in 输入参数介绍:
该计算是在一步能带计算的基础上外加电场,除能带计算的基本参数,新增参数为下:
corr.dipolEfield: 设置外加电场的大小,该参数只在corr.dipol = true和设置corr.dipolDirection的情况下生效;
structure.as 文件参考如下:
1Total number of atoms
22
3Lattice
43.860000 0.000000 0.000000
5-1.930000 3.342860 0.000000
60.000000 0.000000 26.460000
7Direct
8Si 0.333333 0.166667 0.396825
9Si 0.666758 0.833380 0.379216
run程序运行
准备好输入文件之后,将 Efield.in 和 structure.as 文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW Efield.in 。
analysis计算结果分析
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 scf.h5 等输出文件。
scf.h5 :自洽计算对应的 h5 输出文件,当io.band = true时, scf.h5 文件会写入能带数据;
此例中参数 corr.dipolEfield = 0.2 ,即外加电场的大小为 0.2 eV/Å, 在该电场下进行能带计算得到的能带图如图(a)所示,
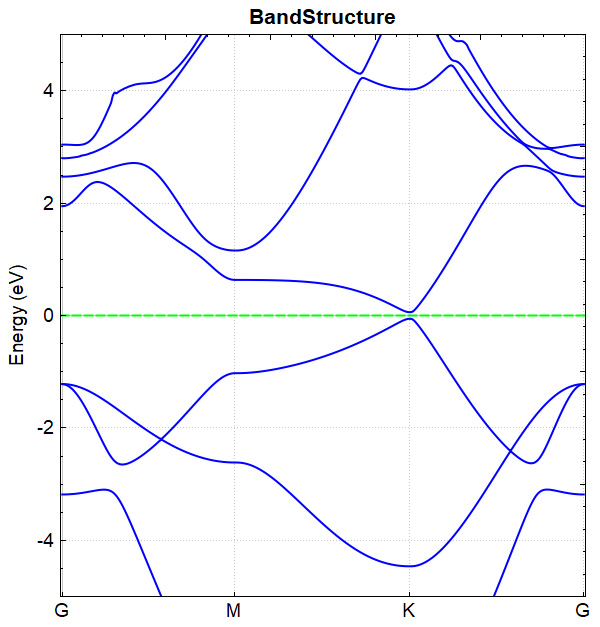
(a)
若设置参数 corr.dipolEfield = 0 重复以上计算,即在无电场的情况下进行能带计算得到的能带图如图(b)所示,
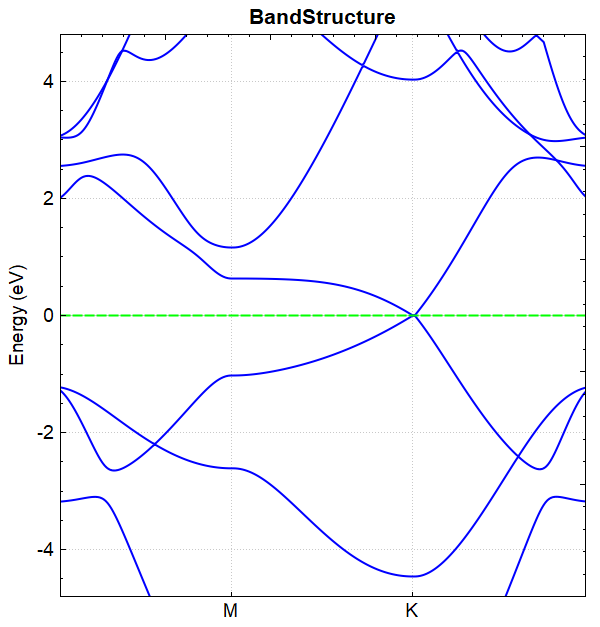
(b)
对比图(a)和图(b)可得结论:通过外加电场可以打开硅烯的带隙。 从 DS-PAW.log 文件可读出加电场与不加电场 BandGap 的值分别为 0.1176 eV 和 0.0010 eV 。
备注
外加电场的单位 eV/Å 为原子受力的单位
2.20 polarization铁电计算
本节将以 \(HfO_{2}\) 为例,介绍在DS-PAW中如何使用现代极化理论进行铁电计算,分析 \(HfO_{2}\) 的铁电极化。
\(HfO_{2}\) 铁电计算输入文件
输入文件包含参数文件 polarization.in 和一系列不同相结构的结构文件 structure.as , polarization.in 如下:
1# task type
2task = scf
3#system related
4sys.structure = structure.as
5sys.symmetry = true
6sys.functional = PBE
7sys.spin = none
8
9#scf related
10cal.methods = 3
11cal.smearing = 4
12cal.cutoff = 520
13cal.ksamping = MP
14cal.kpoints = [4, 4, 4]
15
16scf.convergence = 1e-5
17
18#outputs
19io.charge = false
20io.wave = false
21io.polarization = true
polarization.in 输入参数介绍:
该计算是在自洽计算的基础上进行铁电计算,除自洽计算的基本参数,新增参数为下:
io.polarization: 控制自洽计算中铁电计算的开关;
\(HfO_{2}\) 极化方向向下的铁电相结构 structure.as 文件参考如下:
1Total number of atoms
212
3Lattice
45.04621935 0.00000000 0.00000000
50.00000000 5.07315250 0.00000000
60.00000000 0.00000000 5.25768906
7Cartesian
8Hf 1.34815269 1.22145222 0.17639072
9Hf 1.34815269 3.75802848 2.45245381
10Hf 3.69806665 1.22145222 2.80523525
11Hf 3.69806665 3.75802848 5.08129834
12O 0.35195212 1.93667284 1.92589951
13O 0.35195212 4.47324910 0.70294502
14O 2.32678304 2.48829365 3.85528783
15O 2.32678304 5.02486989 4.03124575
16O 2.71943629 5.02486989 1.40240122
17O 2.71943629 2.48829365 1.22644331
18O 4.69426723 1.93667284 4.55474404
19O 4.69426723 4.47324910 3.33178954

铁电相极化方向向下
\(HfO_{2}\) 极化方向向上的铁电相结构 structure.as 文件参考如下:
1Total number of atoms
212
3Lattice
45.04621935 0.00000000 0.00000000
50.00000000 5.07315250 0.00000000
60.00000000 0.00000000 5.25768906
7Cartesian
8Hf 1.34815269 1.31512402 0.17639072
9Hf 1.34815269 3.85170026 2.45245381
10Hf 3.69806665 1.31512402 2.80523525
11Hf 3.69806665 3.85170026 5.08129834
12O 0.35195212 0.59990340 1.92589951
13O 0.35195212 3.13647965 0.70294502
14O 2.32678304 2.58485884 4.03124575
15O 2.32678304 5.12143510 3.85528783
16O 2.71943630 5.12143510 1.22644331
17O 2.71943630 2.58485884 1.40240122
18O 4.69426723 0.59990340 4.55474404
19O 4.69426723 3.13647965 3.33178954

铁电相极化方向向上
在极化向下和极化向上的结构中插入系列中间过渡结构,使用 线性插值 (neb.linear_interpolate)的方法,具体参见辅助工具 neb_structure.py 脚本,本例插入中间结构 11 个,包括初末态极化相共 13 个构型,对所有构型依次进行极化计算。
run程序运行
准备好输入文件之后,将 polarization.in 和各 structure.as 文件上传到服务器,将13个结构放入13个目录,依次按照结构弛豫中介绍的方法执行 DS-PAW polarization.in
analysis计算结果分析
根据上述的输入文件,计算完成之后将会得到13组 DS-PAW.log 、 scf.h5 和 polarization.txt 等输出文件。
DS-PAW.log :DS-PAW铁电计算之后得到的日志文件;
scf.h5 :自洽计算对应的 h5 输出文件,注意h5文件的名称与task类型严格一致。h5文件解析见 输出文件格式说明 部分;
polarization.txt : 铁电极化计算完成之后的 txt 文本文件,电子、离子贡献的极化部分及总的极化量子数存储在该文件中,便于用户快速获取信息。
以极化向下(00)铁电相体系为例,从 polarization.txt 文件可得 \(HfO_{2}\) 的铁电极化数据如下所示:
Total(x y z) ((μC/cm^2))
-0.000043
-8.715604
-0.000002
Quantum(x y z) (μC/cm^2)
60.067225
60.387821
62.584436
以极化向上(12)铁电相体系为例,从 polarization.txt 文件可得 \(HfO_{2}\) 的铁电极化数据如下所示:
Total(x y z) ((μC/cm^2))
-0.000049
8.715446
0.000001
Quantum(x y z) (μC/cm^2)
60.067225
60.387821
62.584436
可使用 PolaTotal.py 脚本对写入极化数据的 scf.h5 文件进行数据处理,具体操作见 辅助工具使用教程 部分。对13组铁电计算的数据进行处理,得到结果图如下:

13组结构对应极化数值
上图为经过极化量子周期性换算得到的x, y ,z 三个方向的极化强度Px, Py, Pz。因 \(HfO_{2}\) 极化方向为y方向,Px, Pz数值不随原子位移而改变。
2.21 bader电荷计算
本节将以NaCl晶体为例,介绍在DS-PAW中如何进行bader电荷计算,分析NaCl体系中各原子的价态分布。
\(NaCl\) 晶体Bader电荷计算输入文件
输入文件包含参数文件 bader.in 和结构文件 structure.as , bader.in 如下:
bader.in 文件参考如下:
1# task type
2task = scf
3#system related
4sys.structure = structure.as
5sys.symmetry = true
6sys.functional = PBE
7sys.spin = none
8
9#scf related
10cal.methods = 1
11cal.smearing = 1
12cal.ksamping = G
13cal.kpoints = [10, 10, 10]
14cal.cutoff = 650
15#outputs
16io.charge = true
17io.wave = false
18io.bader = true
bader.in 输入参数介绍:
该计算是在自洽计算的基础上进行bader电荷计算,除自洽计算的基本参数,新增参数为下:
io.bader: 控制自洽计算中bader电荷计算的开关;
structure.as 文件参考如下:
1Total number of atoms
28
3Lattice
45.68452692 0.00000000 0.00000000
50.00000000 5.68452692 0.00000000
60.00000000 0.00000000 5.68452692
7Cartesian
8Na 4.26339519 1.42113173 1.42113173
9Na 1.42113173 4.26339519 1.42113173
10Na 1.42113173 1.42113173 4.26339519
11Na 4.26339519 4.26339519 4.26339519
12Cl 1.42113173 1.42113173 1.42113173
13Cl 4.26339519 4.26339519 1.42113173
14Cl 4.26339519 1.42113173 4.26339519
15Cl 1.42113173 4.26339519 4.26339519
备注
io.bader = true 时,io.charge 必须设置为true
run程序运行
准备好输入文件之后,将 bader.in 和 structure.as 文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW bader.in 。
analysis计算结果分析
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 scf.h5 、 bader.txt 等输出文件。
DS-PAW.log :DS-PAW bader电荷计算之后得到的日志文件;
scf.h5 :自洽计算对应的 h5 输出文件,注意h5文件的名称与task类型严格一致。h5文件解析见具体的数据结构详见 输出文件格式说明 部分;
bader.txt : bader电荷计算完成之后的 txt 文本文件,该文件写入bader电荷数据,便于用户快速获取信息。
bader.txt 文本内容如下所示,bader电荷分析得到的数据与utexas大学的Henkelman小组得到的 数据 吻合。
Total number of valence electronics: 64
Element |
X |
Y |
Z |
Charge |
AtomicVolume |
MinDistance |
Cl |
0.25 |
0.25 |
0.25 |
7.85852 |
35.893 |
1.65799 |
Cl |
0.75 |
0.75 |
0.25 |
7.85704 |
35.83 |
1.65799 |
Cl |
0.75 |
0.25 |
0.75 |
7.84024 |
35.0495 |
1.65799 |
Cl |
0.25 |
0.75 |
0.75 |
7.87537 |
36.6765 |
1.65799 |
Na |
0.75 |
0.25 |
0.25 |
8.14221 |
10.0598 |
1.10532 |
Na |
0.25 |
0.75 |
0.25 |
8.14223 |
10.0607 |
1.10532 |
Na |
0.25 |
0.25 |
0.75 |
8.14221 |
10.0598 |
1.10532 |
Na |
0.75 |
0.75 |
0.75 |
8.14221 |
10.0598 |
1.10532 |
2.22 bandunfolding能带反折叠计算
本节将以 \(Cu_{3}Au\) 体系为例,介绍在DS-PAW中如何进行能带反折叠计算,分析 \(Cu_{3}Au\) 反折叠的能带图。
\(Cu_{3}Au\) 能带反折叠计算输入文件
能带反折叠计算需两步法完成能带计算,因此输入文件包含参数文件 scf.in 、 bandunfolding.in 和结构文件 structure.as ,
scf.in 如下:
1task = scf
2
3sys.structure = structure.as
4sys.symmetry = true
5sys.functional = PBE
6sys.spin = none
7
8cal.methods = 1
9cal.smearing = 1
10cal.ksamping = MP
11cal.kpoints = [3, 3, 3]
12cal.cutoff = 650
13
14scf.convergence = 1.0e-05
15
16io.charge = true
17io.wave = false
bandunfolding.in 如下:
1task = band
2cal.iniCharge = ./rho.bin
3
4sys.structure = structure.as
5sys.symmetry = true
6sys.functional = PBE
7sys.spin = none
8
9cal.methods = 1
10cal.smearing = 1
11cal.ksamping = MP
12cal.kpoints = [3, 3, 3]
13cal.cutoff = 500
14
15scf.convergence = 1.0e-05
16
17band.unfolding = true
18band.primitiveUVW=[0.0, 0.5, 0.5, 0.5, 0.0, 0.5, 0.5, 0.5, 0.0]
19band.kpointsLabel= [R,G,X]
20band.kpointsCoord= [0.5, 0.5, 0.5, 0.0, 0.0, 0.0, 0.5, 0.0, 0.5]
21band.kpointsNumber= [101, 101]
22
23io.charge = false
24io.wave = false
bandunfolding.in 输入参数介绍:
能带反折叠计算是在能带计算的基础上完成的,且能带计算必须通过两步法完成。除能带计算的基本参数,新增参数为下:
band.unfolding: 控制能带计算中能带反折叠计算的开关;band.primitiveUVW: 设置UVW系数,超胞的晶格矢量乘上UVW系数等于原胞的晶格矢量,用于控制能带反折叠的参数;
structure.as 文件参考如下:
1Total number of atoms
24
3Lattice
43.7530000210 0.0000000000 0.0000000000
50.0000000000 3.7530000210 0.0000000000
60.0000000000 0.0000000000 3.7530000210
7Direct
8Au 0.000000000 0.000000000 0.000000000
9Cu 0.000000000 0.500000000 0.500000000
10Cu 0.500000000 0.000000000 0.500000000
11Cu 0.500000000 0.500000000 0.000000000
run程序运行
准备好输入文件之后,将 scf.in 、 bandunfolding.in 和 structure.as 文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW scf.in ,自洽计算完成后执行 DS-PAW bandunfolding.in 。
analysis计算结果分析
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 scf.h5 、 band.h5 等输出文件。
band.h5 :能带计算对应的 h5 输出文件,相比能带计算该文件新增 UnfoldingBandInfo 部分,具体结构解析见 输出文件格式说明 部分。
可使用脚本 bandunfolding.py 对 band.h5 进行数据处理,具体操作见 辅助工具使用教程 部分。该例得到的能带效果图应如下所示,与文献报道结果[2] 一致。
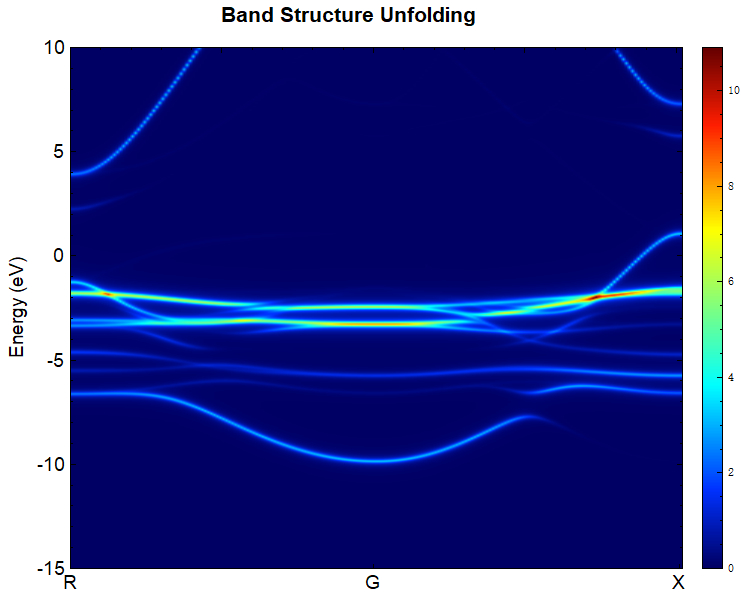
2.23 epsilon介电常数计算
本节将以Si体系为例,介绍在DS-PAW中如何进行介电常数计算。
\(Si\) 介电常数计算输入文件
输入文件包含参数文件 epsilon.in 和结构文件 structure.as , epsilon.in 如下:
1# task type
2task = epsilon
3#system related
4sys.structure = structure.as
5sys.symmetry = true
6sys.functional = PBE
7sys.spin = none
8
9#scf related
10cal.methods = 1
11cal.smearing = 1
12cal.ksamping = G
13cal.kpoints = [5, 5, 5]
14cal.cutoff = 500
15scf.convergence = 1.0e-7
epsilon.in 输入参数介绍:
介电常数的计算可通过直接指定 task 完成,新增task的可选值如下:
task: 设置计算类型,新增 epsilon 参数,此处对应介电常数的计算;
备注
在 task = phonon 且 phonon.method = dfpt 时也可完成介电常数的计算,通过添加 phonon.dfptEpsilon = true 参数即可。
structure.as 文件参考如下:
1Total number of atoms
28
3Lattice
4 5.43070000 0.00000000 0.00000000
5 0.00000000 5.43070000 0.00000000
6 0.00000000 0.00000000 5.43070000
7Cartesian
8Si 0.67883750 0.67883750 0.67883750
9Si 3.39418750 3.39418750 0.67883750
10Si 3.39418750 0.67883750 3.39418750
11Si 0.67883750 3.39418750 3.39418750
12Si 2.03651250 2.03651250 2.03651250
13Si 4.75186250 4.75186250 2.03651250
14Si 4.75186250 2.03651250 4.75186250
15Si 2.03651250 4.75186250 4.75186250
run程序运行
准备好输入文件之后,将 epsilon.in 和 structure.as 文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW epsilon.in 。
analysis计算结果分析
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 epsilon.h5 、 epsilon.txt 等输出文件。
DS-PAW.log :DS-PAW介电常数计算之后得到的日志文件;
epsilon.h5 :介电常数计算对应的 h5 输出文件,具体的数据结构详见 输出文件格式说明 部分;
epsilon.txt : 介电常数计算完成之后的 txt 文本文件,该文件写入介电常数相关数据,便于用户快速获取信息。
从 epsilon.txt 文件中可获取如下数据:
Total Part |
||
13.309902 |
0.000000 |
-0.000000 |
-0.000000 |
13.309902 |
-0.000000 |
-0.000000 |
0.000000 |
13.309902 |
分析上表可得该体系的介电常数为 13.309902 ,与文献报道值[3] 13.31 一致。
2.24 piezo压电张量计算
本节将以AlN体系为例,介绍在DS-PAW中如何进行压电张量计算,得到材料的压电系数 \(e_{33} (0)\)。
\(AlN\) 压电张量计算输入文件
输入文件包含参数文件 piezo.in 和结构文件 structure.as , piezo.in 如下:
1task = epsilon
2#system related
3sys.structure = structure.as
4sys.symmetry = true
5sys.functional = PBE
6sys.spin = none
7
8#scf related
9cal.methods = 1
10cal.smearing = 1
11cal.ksamping = G
12cal.kpoints = [10, 10, 10]
13cal.cutoffFactor = 1.5
14scf.convergence = 1.0e-7
15
16#outputs
17io.charge = false
18io.wave = false
piezo.in 输入参数介绍:
task: 设置计算类型,新增 epsilon 参数,此处对应压电张量计算;scf.convergence: 设置介电张量计算电子收敛的精度,建议提高精度,此处设置为1.0e-7;
structure.as 文件参考如下:
1Total number of atoms
28
3Lattice
43.11606630 0.00000000 0.00000000
50.00000000 5.39683518 0.00000000
60.00000000 0.00000000 5.00770902
7Cartesian
8Al 0.00000000 3.59735137 0.00946380
9Al 0.00000000 1.79945276 2.51320124
10Al 1.55803315 0.89899597 0.00945662
11Al 1.55803315 4.49786165 2.51308138
12N 0.00000000 3.59851112 1.91845914
13N 0.00000000 1.79831356 4.42266820
14N 1.55803315 0.90013952 1.91851680
15N 1.55803315 4.49672497 4.42258192
run程序运行
准备好输入文件之后,将 piezo.in 和 structure.as 文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW piezo.in 。
analysis计算结果分析
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 epsilon.h5 、 epsilon.txt 等输出文件。
DS-PAW.log :DS-PAW压电张量计算之后得到的日志文件;
epsilon.h5 :介电常量计算对应的 h5 输出文件,具体的数据结构详见 输出文件格式说明 部分;
epsilon.txt : 压电计算完成之后的 txt 文本文件,该文件写入压电相关数据,便于用户快速获取信息。
从 epsilon.txt 文件中可获取如下数据:
Piezoelectric Tensor (C/m^2)( Row: x y z Column: XX YY ZZ XY YZ ZX) |
|||||
Electronic Part |
|||||
0.000000 |
0.000000 |
0.000000 |
0.000006 |
0.000000 |
0.336610 |
-0.000001 |
0.000007 |
0.000003 |
0.000000 |
0.336662 |
0.000000 |
0.266339 |
0.265888 |
-0.419569 |
0.000000 |
-0.000014 |
0.000000 |
Ionic Part: |
|||||
-0.000004 |
0.000002 |
0.000002 |
0.000032 |
-0.000000 |
-0.681702 |
-0.000163 |
-0.000239 |
0.000314 |
-0.000000 |
-0.699012 |
-0.000000 |
-0.911456 |
-0.913265 |
1.943887 |
-0.000000 |
-0.000633 |
-0.000000 |
Total Part: |
|||||
-0.000004 |
0.000002 |
0.000002 |
0.000039 |
-0.000000 |
-0.345092 |
-0.000164 |
-0.000232 |
0.000317 |
-0.000000 |
-0.362350 |
-0.000000 |
-0.645117 |
-0.647377 |
1.524318 |
-0.000000 |
-0.000647 |
-0.000000 |
分析上表可得压电张量电子贡献部分 \(e_{33} (0)\) 的数值为 -0.419569 \(C/m^{2}\) ,总的压电张量 \(e_{33}\) 的数值为 1.524318 \(C/m^{2}\) ,与文献参考值[4] -0.47 \(C/m^{2}\) 和 1.46 \(C/m^{2}\) 接近。
2.25 fixcell固定基矢弛豫计算
本节将以 \(MoS_{2}\) 体系为例,介绍在DS-PAW中如何进行固定晶格弛豫计算。
\(MoS_{2}\) 固定基矢弛豫计算输入文件
输入文件包含参数文件 relax.in 和结构文件 structure.as , relax.in 如下:
1# task type
2task = relax
3#system related
4sys.structure = structure.as
5sys.symmetry = false
6sys.functional = PBE
7sys.spin = none
8
9#scf related
10cal.methods = 1
11cal.smearing = 1
12cal.ksamping = G
13cal.cutoff = 650
14cal.kpoints = [19, 19, 5]
15#relax related
16relax.freedom = all
17relax.convergence = 0.05
18relax.methods = CG
structure.as 文件参考如下:
1Total number of atoms
26
3Lattice Fix_x Fix_y Fix_z
43.19031572 0.00000000 0.00000000 F T T
5-1.59515786 2.76289446 0.00000000 F F T
60.00000000 0.00000000 14.87900448 T T T
7Cartesian
8S 0.00000000 1.84193052 12.72413785
9S 1.59515943 0.92096386 5.28463561
10S 0.00000000 1.84193052 9.59436887
11S 1.59515943 0.92096386 2.15486663
12Mo 1.59515943 0.92096386 11.15925336
13Mo 0.00000000 1.84193052 3.71975112
structure.as 标签设置介绍:
固定晶胞维度进行弛豫计算需在 structure.as 文件中新增固定标签,类似于固定原子弛豫的设置(在原子坐标后添加Fix标签),固定基矢需在 structure.as 的第三行 Lattice 后添加Fix标签,本案例的标签对应固定晶胞的c边和a边的y、z方向、b边的z方向。
run程序运行
准备好输入文件之后,将 relax.in 和 structure.as 文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW relax.in 。
analysis计算结果分析
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 relax.h5 、 latestStructure.as 等输出文件。
relax.h5 :弛豫计算对应的 h5 输出文件;
latestStructure.as :弛豫终点的as结构文件,可直接查看数据;
将 latestStructure.as 拖入Device Studio查看结构,或直接打开该文件,可得弛豫结束后的结构数据如下:
1Total number of atoms
26
3Lattice
43.19696732 0.00000000 0.00000000
5-1.59848077 2.76865753 0.00000000
60.00000000 0.00000000 14.87900448
7Direct
8Mo 0.66666701 0.33333316 0.74999995
9Mo 0.33333340 0.66666675 0.24999997
10S 0.33333340 0.66666666 0.85535854
11S 0.66666686 0.33333303 0.35535875
12S 0.33333367 0.66666699 0.64464148
13S 0.66666708 0.33333333 0.14464130
通过对比可得弛豫前 a = b = 3.19031572 ,弛豫后变成了a = b = 3.19696732 ,而c = 14.87900448 不变。
2.26 thermal声子热力学性质计算
本节将以Si体系为例,介绍在DS-PAW中如何进行声子热力学性质计算。
\(Si\) 声子热力学性质计算输入文件
输入文件包含参数文件 phonon-thermal.in 和结构文件 structure.as , phonon-thermal.in 如下:
1# task type
2task = phonon
3#system related
4sys.structure = structure.as
5sys.symmetry = true
6sys.functional = PBE
7sys.spin = none
8
9#scf related
10cal.methods = 1
11cal.smearing = 1
12cal.ksamping = G
13cal.kpoints = [5, 5, 5]
14cal.cutoffFactor = 1.5
15scf.convergence = 1.0e-7
16#phonon related
17phonon.structureSize = [2,2,2]
18phonon.type =dos
19phonon.qpoints = [31,31,31]
20phonon.method = dfpt
21
22phonon.thermal=true
23phonon.thermalRange = [0,1000,10]
phonon-thermal.in 输入参数介绍:
phonon.thermal: 控制声子计算中热力学计算的开关,仅在phonon.method = dfpt时生效;phonon.thermalRange: 设置热力学计算的温度范围及数据存储间隔;
structure.as 文件参考如下:
1Total number of atoms
22
3Lattice
40.00 2.75 2.75
52.75 0.00 2.75
62.75 2.75 0.00
7Direct
8Si -0.125000000 -0.125000000 -0.125000000
9Si 0.125000000 0.125000000 0.125000000
run程序运行
准备好输入文件之后,将 phonon-thermal.in 和 structure.as 文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW phonon-thermal.in 。
analysis计算结果分析
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 phonon.h5 等输出文件。
DS-PAW.log :DS-PAW声子计算得到的日志文件;
phonon.h5 :DS-PAW声子计算对应的 h5 输出文件,打开热力学计算的开关,生成的 phonon.h5 文件中会写入 ThermalInfo 数据,具体解析见 输出文件格式说明 部分。
可使用 phonon_thermal.py 脚本对声子热力学数据进行处理,具体操作见 辅助工具使用教程 部分。分析热力学数据,可得熵、热容、亥姆霍兹自由能随温度变化的曲线如下所示,与phonony git仓库展示的 结果 一致:

2.27 solid state NEB计算
本节将以HfZrO体系为例,介绍在DS-PAW中如何放开晶胞弛豫进行solid state NEB计算。
\(HfZrO\) Solid state NEB计算输入文件
输入文件包含参数文件 ssneb.in 和结构文件 structure.as , ssneb.in 如下:
1task = neb
2
3sys.structure = structure.as
4sys.functional = LDA
5sys.spin = none
6sys.symmetry = false
7
8cal.ksamping = G
9cal.kpoints = [10,10,10]
10cal.cutoff = 650
11cal.methods = 1
12cal.smearing = 1
13cal.sigma = 0.05
14
15scf.mixType = Broyden
16scf.mixBeta = 0.4
17scf.convergence = 1e-6
18scf.max = 300
19
20neb.springK = 5
21neb.images = 6
22neb.iniFin = true
23neb.method = QM2
24neb.convergence = 0.01
25neb.max = 500
26neb.freedom = all
27
28io.wave = false
29io.charge = false
ssneb.in 输入参数介绍:
neb.freedom: 设置过渡态弛豫的维度,设置为all对应弛豫晶胞大小;neb.method: 设置过渡态搜寻的方法,当 neb.freedom = all 时该参数可选值为QM2和FIRE;
structure.as 需提供多个,初态结构 structure00.as 参考如下
1Total number of atoms
212
3Lattice
45.00209138 0.00000009 0.00000004
50.00000009 5.00209143 -0.00000004
60.00000004 -0.00000004 5.07896990
7Cartesian
8Hf 2.50104558 2.50104575 0.00000000
9Hf 0.00000000 0.00000000 0.00000000
10O 3.75156841 1.25052303 1.47285183
11O 3.75156857 3.75156869 1.04735062
12O 1.25052293 1.25052297 3.60611823
13O 1.25052286 3.75156867 4.03161932
14O 1.25052287 3.75156860 1.47285187
15O 1.25052275 1.25052294 1.04735054
16O 3.75156850 1.25052287 4.03161945
17O 3.75156850 3.75156869 3.60611821
18Zr 2.50104577 0.00000000 2.53948497
19Zr 0.00000000 2.50104594 2.53948491
末态结构 structure07.as 参考如下
1Total number of atoms
212
3Lattice
44.98221520 -0.00002552 0.00036684
5-0.00002562 4.99587652 0.00005905
60.00039053 0.00006126 5.18258321
7Cartesian
8Hf 2.30823006 2.49975412 0.04967381
9Hf 0.00919001 0.00195723 0.38722458
10O 4.03365086 0.66419181 2.12958714
11O 4.00001549 3.18954023 0.89210846
12O 0.95871628 1.24120307 4.04442128
13O 0.94984693 3.74053908 4.19050825
14O 1.35895285 3.73907584 1.57483409
15O 1.36804279 1.24264997 1.42944278
16O 3.29999107 0.69159253 4.72728663
17O 3.26626721 3.16200890 3.48972595
18Zr 2.31915914 0.00841995 2.97686955
19Zr 4.98082249 2.50639160 2.64290889
初末态构型在 Device Studio 中显示如下:
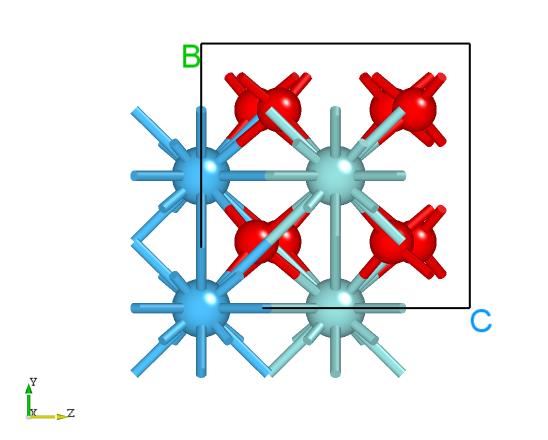
初态T相构型
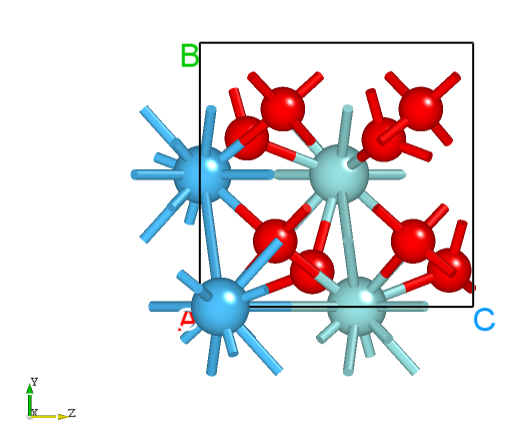
末态F相构型
备注
在
neb.freedom = all时,neb.method可选值为 QM2 或 FIRE中间结构的生成可调用“辅助工具使用教程-过渡态部分”的neb_interpolate_structures.py脚本,完成插值可调用neb_visualize.py脚本对插值结构进行预览,可调用calc_dist.py脚本查看image之间的距离是否合理。
run程序运行
准备好输入文件之后,将 ssneb.in 和包含 structureNo.as 文件的多个文件夹上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW ssneb.in 。
analysis计算结果分析
根据上述的输入文件,计算完成之后:
>> 初态和末态结构所在文件夹会生成自洽计算所得的 DS-PAW.log 、latestStructure00.as 、 scf.h5 等输出文件;
>> 中间结构 structureNo.as 所在文件夹 No (参与过渡态计算的中间结构所在文件夹,中间结构的个数由 neb.images 参数决定)会生成结构优化所得的 nebNo.h5 、 latestStructureNo.as 等输出文件;
>> 最外层目录将会生成 DS-PAW.log 、 neb.h5 这2个文件,其中 neb.h5 为 No 文件夹下各 nebNo.h5 文件的信息汇总。
DS-PAW.log :DS-PAW过渡态计算之后得到的日志文件;
neb.h5 :过渡态计算完成之后的 h5 数据文件;此时反应坐标及能量变化等数据被保存在 neb.h5 中,具体的数据结构详见 输出文件格式说明 部分;
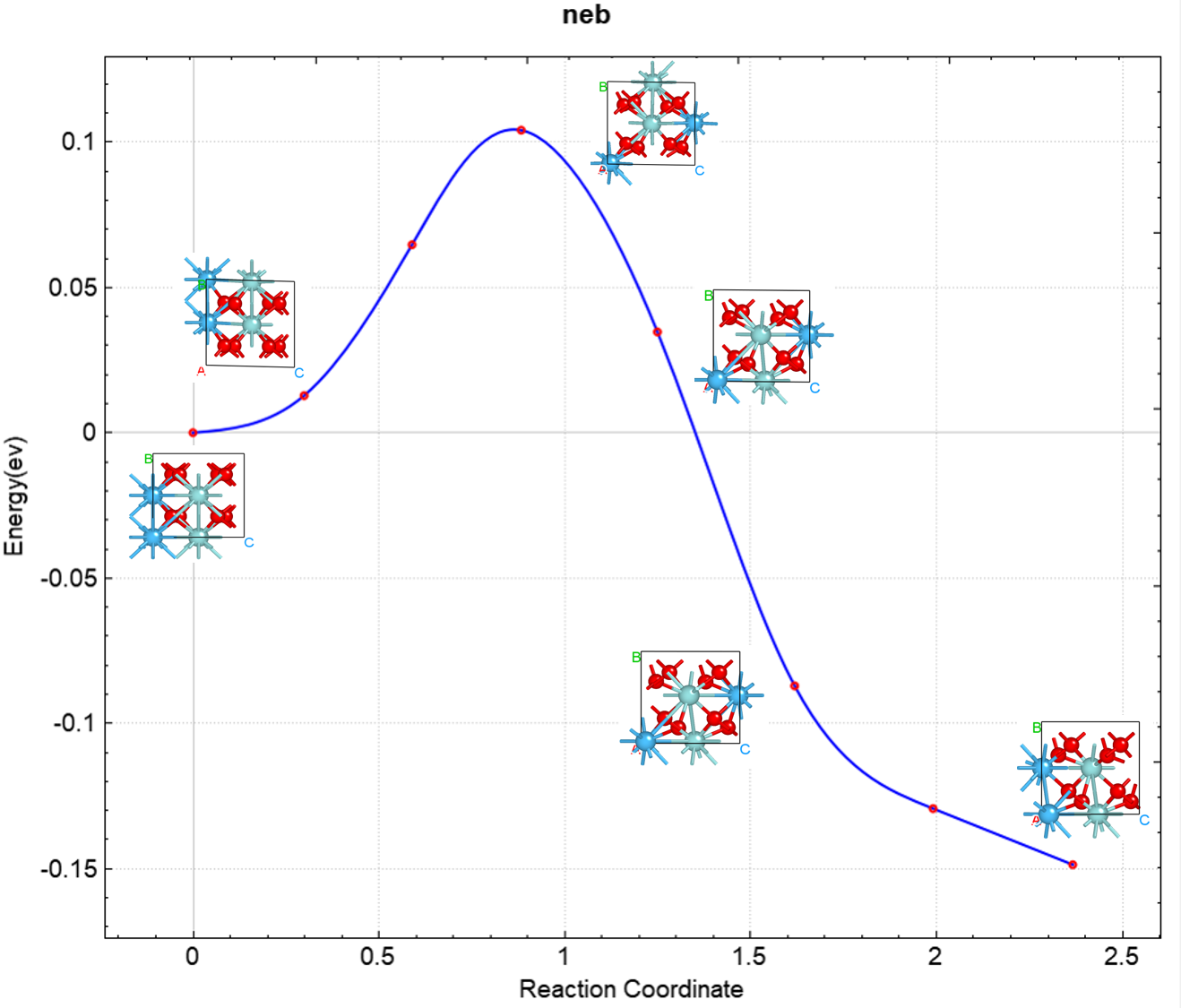
可使用 python 脚本 neb.py 对neb计算的结果进行分析,需在完整的neb计算目录下执行分析脚本,具体操作见 辅助工具使用教程 部分。处理得到的反应势垒曲线效果应如下所示:
2.28 solvation溶剂化能计算
本节将以 \(H_{2}O\) 体系为例,介绍在DS-PAW中如何计算隐式溶剂模型下的溶剂化能。
\(H_{2}O\) 溶剂化能计算输入文件
输入文件包含参数文件 scf.in 和结构文件 structure.as , scf.in 如下:
1# task type
2task = scf
3#system related
4sys.structure = structure.as
5sys.symmetry = true
6sys.functional = PBE
7sys.spin = none
8
9#scf related
10cal.methods = 1
11cal.smearing = 3
12cal.sigma = 0.2
13cal.ksamping = G
14cal.kpoints = [1, 1, 1]
15cal.supGrid = true
16cal.cutoff = 800
17scf.convergence = 1.0e-6
18
19#implicit solvation model
20sys.sol = true
21sys.solEpsilon = 80
22sys.solTAU = 5.25E-4
23
24#outputs
25io.charge = false
26io.wave = false
27io.boundCharge = true
scf.in 输入参数介绍:
sys.sol: 控制引入隐式溶剂化模型的开关,true则考虑溶剂化效应;sys.solEpsilon: 设置溶剂介电常数大小,此例设置为80;sys.solTAU: 指定单位面积的有效界面张力的大小,单位eV/Å^2,默认值为5.25E-4,该参数建议设置为小于1e-3的数值;io.boundCharge: 控制溶剂束缚电荷密度文件输出的开关。
结构文件 structure.as 参考如下
1Total number of atoms
23
3Lattice
410.00000000 0.00000000 0.00000000
50.00000000 10.00000000 0.00000000
60.00000000 0.00000000 10.00000000
7Cartesian
8H 5.63934499 4.89541998 4.58224001
9H 4.36065501 5.11499002 5.45934003
10O 4.65002501 4.88500998 4.54065997
run程序运行
准备好输入文件之后,将 scf.in 和 structure.as 文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW scf.in 。
analysis计算结果分析
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 scf.h5 、 rhoBound.h5 等输出文件。
DS-PAW.log :DS-PAW隐式溶剂化模型计算之后得到的日志文件;
scf.h5 :自洽计算对应的 h5 输出文件;
rhoBound.h5 :使用隐性溶剂模型计算得到的溶剂束缚电荷密度文件;
此例计算可得到考虑溶剂化能的总能 \(E_{sol}\) ,溶剂化能计算公式如下:
Solvation Energy=E(sys.sol=true)-E(sys.sol=false)
根据该公式可知,需另进行sys.sol = false的计算以获得不考虑溶剂化能的总能 \(E_{nosol}\),将 \(E_{nosol}\) 代入以上公式可得水的溶剂化能为 -0.313 eV,与文献[5] 报道的结果一致。
使用隐式溶剂化模型进行计算时,可以同时得到溶质周围的溶剂束缚电荷密度分布文件 rhoBound.h5 ,可使用 python 脚本 trans_rho.py 对该文件进行后处理,具体操作见 辅助工具使用教程 部分。转换成的可视化文件在VESTA中打开,可以得到如下等密度面分布图:
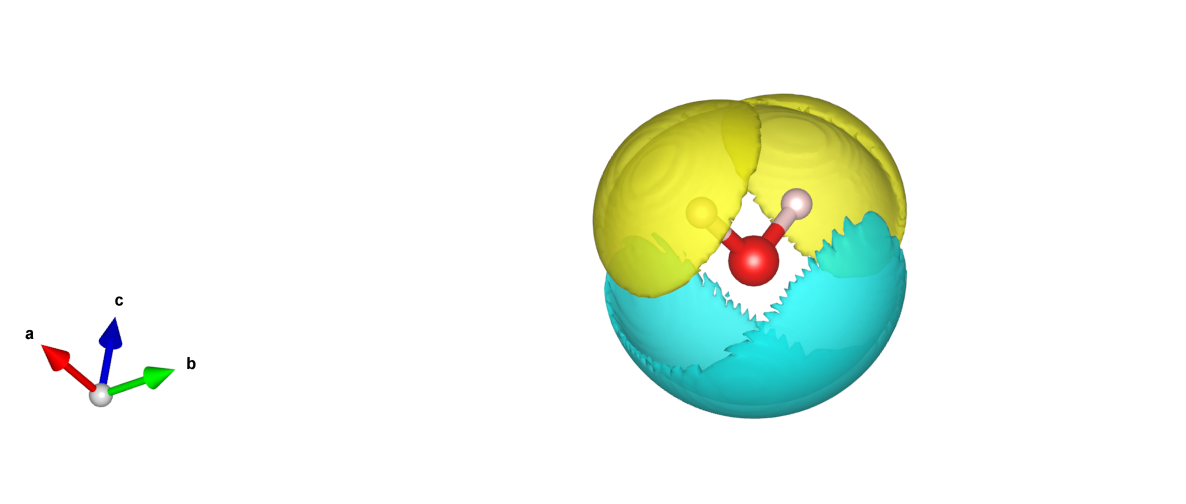
从图中可以看到,溶剂化的正负屏蔽电荷密度分布在水分子的外围,形成一个溶解壳层,符合模型的预期,并与其它软件算得的溶剂束缚电荷密度分布类似。
2.29 fixedpotential固定电势计算
本节将以 \(Cu-slab\) 体系为例,介绍如何在DS-PAW中进行固定电势计算。
\(Cu-slab\) 固定电势计算输入文件
输入文件包含参数文件 fixedP.in 和结构文件 structure.as , fixedP.in 如下:
1# task type
2task = scf
3
4sys.functional = PBE
5sys.structure = structure.as
6
7cal.ksamping = G
8cal.cutoff = 650
9cal.sigma = 0.2
10cal.smearing = 3
11cal.kpoints = [7,7,1]
12
13scf.convergence = 1.0e-6
14scf.max = 200
15
16sys.sol = true
17sys.solEpsilon = 78.4
18sys.solLambdaD = 3.04
19sys.solTAU = 0
20
21# Potential fixed
22sys.fixedP = true
23sys.fixedPPotential = 2.155
24
25io.charge = true
26io.wave = false
fixedP.in 部分输入参数介绍:
task: 设置计算类型,本例在task=scf时作固定电势计算;sys.sol: 打开溶剂化模型,固定电势计算需在隐式溶剂模型的基础上完成;sys.solEpsilon: 设置溶剂介电常数大小,此例设置为 78.4;sys.solLambdaD: 使用泊松玻尔兹曼方程且设置德拜长度(Debye length)的值,若不设置,则使用的是泊松方程,没有描述界面离子对静电势的贡献;sys.solTAU: 指定单位面积的有效界面张力的大小,单位eV/Å^2,默认值为5.25E-4,该参数建议设置为小于1e-3的数值;sys.fixedP: 打开固定电势计算的开关;sys.fixedPPotential: 设置固定电势的电势值,默认以标准氢电极电势(SHE)作为参考电极电势,若以零电荷电位(Potential of Zero Charge, PZC)作为参考电极可设置参数sys.fixedPType = PZC;
备注
关于德拜长度
sys.solLambdaD, 其表达式为 \(\lambda_D = \sqrt\frac{\varepsilon \varepsilon_ok_B T}{2 c^0 z^2 q^2}\)
1M 带+/-1电荷阴阳离子的水溶液的德拜长度为:3.04 Å
structure.as 文件参考如下:
1Total number of atoms
28
3Lattice
43.63404989 0.00000000 0.00000000
50.00000000 3.63404989 0.00000000
60.00000000 0.00000000 23.62132454
7Cartesian
8Cu 0.00000000 0.00000000 1.81702310
9Cu 1.81702495 0.00000000 3.63404620
10Cu 1.81702495 1.81702495 1.81702310
11Cu 0.00000000 1.81702495 3.63404620
12Cu 0.00000000 0.00000000 5.46390548
13Cu 1.81702495 0.00000000 7.22885308
14Cu 1.81702495 1.81702495 5.46390548
15Cu 0.00000000 1.81702495 7.22885308
run程序运行
准备好输入文件之后,将 fixedP.in 和 structure.as 文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW fixedP.in 。
analysis计算结果分析
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 scf.h5 等输出文件。
DS-PAW.log :DS-PAW完成固定电势计算之后得到的日志文件;
scf.h5 :task等于scf时DS-PAW对应的 h5 输出文件;具体的数据结构详见 输出文件格式说明 部分;
DS-PAW 采用最速下降法进行固定电势计算,计算过程中迭代体系的电荷数进行多次自洽计算。 DS-PAW.log 文件会写入多次自洽计算的收敛过程,此例在 LOOP 5 达到收敛精度,如下展示 LOOP 5 结束体系对应的电势数值:
1## FINISHED FIXEDPOTENTIAL LOOP 5 ##
2Electron : 149.993000
3ElectrodePotential_SHE : 2.157747 V
4ElectrodePotential_PZC : 2.484286 V
5ElectrodePotential_SHE(PZC) : -0.326539 V
6Chemical Potential(electron) : -6.757747 eV
7Grand Total Energy(sigma->0) : -43088.518081 eV
其中
计算结束所得体系的电势值为 2.157 V,与目标电势值 2.155 V 接近。
备注
进行固定电势计算时必须在隐式溶剂模型下进行,即 sys.fixedP = true 时,必须设置 sys.sol = true 。
目前,仅在 task = scf 时支持固定电势计算。
ElectrodePotential_SHE=ElectrodePotential_PZC+ElectrodePotential_SHE(PZC)
2.30 wannier插值能带计算
本节将以 \(Si\) 体系为例,介绍如何在DS-PAW中使用wannier函数进行插值能带计算。
\(Si\) 插值能带计算输入文件
输入文件包含参数文件 wannier.in 和结构文件 structure.as , wannier.in 如下:
1# task type
2task = wannier
3sys.structure = structure.as
4sys.symmetry = false
5sys.functional = PBE
6sys.spin = none
7cal.methods = 1
8cal.smearing = 1
9cal.ksamping = G
10cal.kpoints = [16,16,16]
11cal.totalBands = 12
12
13#wannier related
14wannier.functions = 12
15wannier.wannMaxIter = 20000
16wannier.outStep = 50
17
18#interpolated band related
19wannier.interpolatedBand = true
20wannier.kpointsLabel= [G,X,W,K,G,L]
21wannier.kpointsCoord= [0, 0, 0, 0.5, 0, 0.5, 0.5, 0.25, 0.75, 0.375, 0.375, 0.75, 0, 0, 0, 0.5, 0.5, 0.5]
22wannier.kpointsNumber = [100]
23
24io.charge = true
25io.wave = true
wannier.in 输入参数介绍:
task = wannier: 设置计算类型,新增可选值wannier,用于进行wannier计算;wannier.functions: 设置wannier函数的个数;wannier.wannMaxIter: 设置求解最大局域化wannier函数过程中的总迭代次数;wannier.outStep:设置task=wannier时输出文件中输出迭代结果的步长;wannier.interpolatedBand: 控制插值能带计算的开关;wannier.kpointsLabel: 设置wannier函数拟合插值能带时高对称点符号;wannier.kpointsCoord: 设置wannier函数拟合插值能带时高对称点坐标;wannier.kpointsNumber: 设置插值能带高对称 K 点之间的撒点数;此例设置参数为 wannier.kpointsNumber= [100] ,高对称点G与X之间撒点数为100,以此求得撒点密度;对高对称点X与W、W与K、K与G、G与L之间进行等密度撒点,实际撒点数可从DS-PAW.log的参数打印部分获取;
structure.as 文件参考如下:
1Total number of atoms
22
3Lattice
40.00 2.75 2.75
52.75 0.00 2.75
62.75 2.75 0.00
7Direct
8Si -0.125000000 -0.125000000 -0.125000000
9Si 0.125000000 0.125000000 0.125000000
备注
初始投影的设置在 structure.as 文件中完成,首先在第7行添加 WannProj 标签,然后在原子坐标后写入初始投影轨道名称, DS-PAW 可识别的投影轨道名称见 参数说明 的 wannier 部分。
此例未自定义初始投影,程序执行随机选择初始投影。若需定义初始投影,可参考下述写法。
自定义初始投影轨道的 structure.as 文件参考如下:
1Total number of atoms
22
3Lattice
40.00 2.75 2.75
52.75 0.00 2.75
62.75 2.75 0.00
7Direct WannProj
8Si -0.125000000 -0.125000000 -0.125000000 [s,p,sp3-1,sp3-2]
9Si 0.125000000 0.125000000 0.125000000 [s,p,sp3-3,sp3-4]
备注
自定义初始投影轨道时,structure.as 文件中投影轨道总数需等于wannier函数的个数(wannier.functions),否则程序报错。
此例总投影轨道为 2*(1+3+1+1) = 12,与 wannier.functions = 12 参数设置一致。
对无需设置初始投影轨道的原子,在对应坐标后写入 [] 即可。
此例cal.totalBands设置为12,因此提交计算时需调用12核。
run程序运行
准备好输入文件之后,将 wannier.in 和 structure.as 文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW wannier.in 。
analysis计算结果分析
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 wannier.h5 等输出文件。
DS-PAW.log :DS-PAW完成wannier插值能带计算之后得到的日志文件;
wannier.h5 :wannier函数插值能带计算对应的 h5 输出文件;具体的数据结构详见 输出文件格式说明 部分;
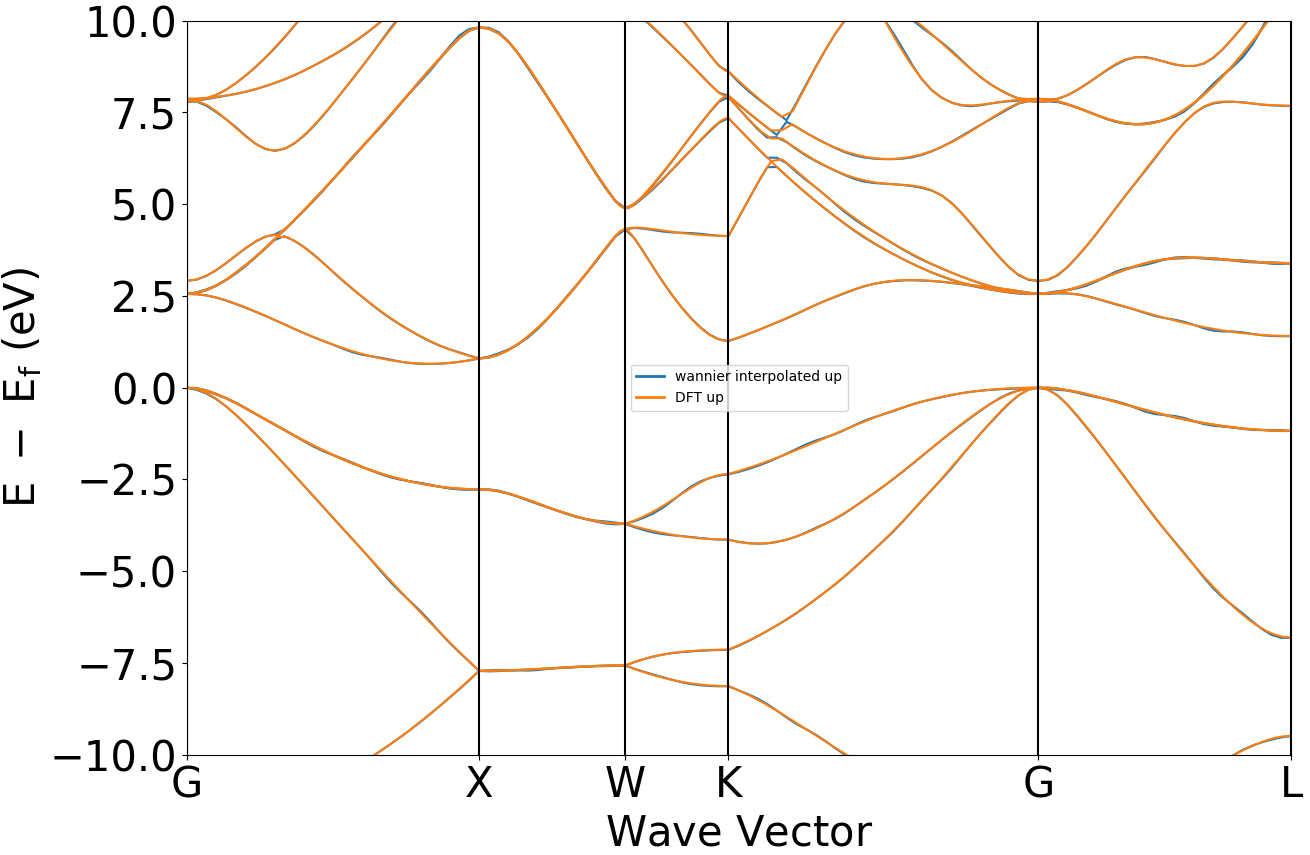
可使用 辅助工具使用教程 –> band能带数据处理 –> bandplot.py 脚本直接对wannier插值能带作图,读取 wannier.h5 即可。
也可使用 bandcompare.py 对wannier插值能带与DFT能带图对比,具体操作见 辅助工具使用教程 部分。能带对比效果图应如下所示:
备注
wannier 计算不支持打开 pob ,且dft能带计算数(cal.totalBands)会随程序调用核数(cores)而改变,因此建议 wannier 计算调用 cores 与 cal.totalBands 参数保持一致。
当 wannier 函数的个数(wannier.functions)小于 cal.totalBands 时,wannier 函数最大局域化的过程需解纠缠,此时若用户未定义能量冻结窗口(wannier.disFrozWin),程序执行提取默认Frozen Window进行计算。
若自定义Frozen Window,需确保wannier.FrozWin 内所含能带的数量不能大于 wannier.functions 的数量,否则程序会报错 E4024。同时需确保窗口的合理性,否则无法得到好的拟合结果。
2023A版本 DS-PAW 暂不支持 spin 类型为 non-collinear 的wannier计算。